

Copyrights: Samaila Musa Chiroma, Mohamad Taufik Hidayat Baharuldin, Che Norma Mat Taib, Zulkhairi Amom, Saravanan Jagadeesan, Mohamad Aris Mohd Moklas, 2018. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Background: There is a dearth of precise information for molecular and cellular mechanisms responsible for the development of Alzheimer’s disease (AD). However, convincing data from clinical research and basic molecular biology have shown that inflammation of the brain is an integral part of AD. In this review, the role of inflammation in AD will be highlighted.
Methods: Articles from credible scientific databases, such as ScienceDirect, Scopus, PubMed, Google Scholar and Mendeley, were searched and retrieved using keywords ‘inflammation’, ‘Alzheimer’s disease’, ‘tau’, and ‘beta amyloid’.
Results: At present, there is no local inflammatory-inciting factor that is closely associated with AD, although it has been proposed that inflammation could be induced by pathologic hallmarks of AD, such as beta amyloid (Aβ) peptide plagues and neurofibrillary tangles (NFTs), or fragments of degenerated neurons. However, it is still unclear whether inflammation leads to the development of AD or if the pathological hallmarks of AD induce inflammation.
Conclusion: Inflammation is, indeed, an integral part of AD. Further studies on inflammatory-targeted therapies for AD are highly recommended.
Introduction
Alzheimer’s disease (AD), the commonest cause of dementia among the elderly people, is an irreversible and progressive neurodegenerative disorder that slowly destroys memory, thinking skills, cognitive functions, and subsequently, the ability to carry out day-to-day activities 1. With the current rise of the aging population in both developed and developing countries, AD has taken center stage as a global health challenge 23. The presence of intracellular neurofibrillary tangles and extracellular beta amyloid (Aβ) plaques in the brain are the two cardinal pathological hallmarks of AD, even though several other hypotheses on the pathogenesis of AD have been suggested and tested, such as neuronal loss, axonal injury, tau phosphorylation, and disturbance of the neurotransmitter acetylcholine 4.
The amyloid beta hypothesis of AD is a highly favored one and the most commonly applied hypothesis in the field of AD research. It has provided the rationale for the development of various treatment modalities. The principle behind this hypothesis is that Aβ peptides produced by the amyloid precursor protein (APP) are the main culprits in AD pathogenesis 56. Based on this hypothesis, several mechanisms for Aβ generation and deposition in the brain have been suggested. These include mitochondrial dysfunction, oxidative stress, aging, apoptosis, genetic factors and inflammatory processes 1. Furthermore, other conditions that have been considered as likely risk factors for developing AD; these include stroke, diabetes, cardiovascular diseases, gender, lower level of education and traumatic brain injury 789101112.
Inflammation is considered to be a major factor in AD whereby the neurofibrillary tangles and beta amyloid plaques co-localize with astrocytes and microglia as well as local immune cells in the brain 13. According to epidemiological studies, patients placed on long-term anti-inflammatory drugs have a reduced risk of developing AD. Meanwhile, genome-wide association studies (GWAS) have revealed a robust relationship between immunological function-regulating genes and AD 141516. Substantiating these observations, both tau and amyloid pathologies have been shown to be induced by pro-inflammatory stimuli 1718.
To date, numerous trials for AD have been carried out with different anti-inflammatory drugs. Both non-steroidal anti-inflammatory drugs (NSAIDs) and steroidal drugs were used for the trials but none of these drugs demonstrated a clear clinical effectiveness in the treatment of AD patients 19. Since the pathogenesis of AD develops long before the manifestation of the clinical symptoms it is, therefore, suggested that anti-inflammatory agents should be administered prodromal 20. Despite the fact that the largest trial conducted on the preventive effect of NSAIDs on AD has failed 20, a recent review of the literature shows support for the use of NSAIDs for preventing AD 21.
The fundamental mechanisms that implicate immune responses in AD pathology are still not clear because the inflammation process could have either a detrimental or beneficial 22 effect on AD, depending on the stage of the disease 23. An example of pro-inflammatory mediators secreted from active immune cells are microglia, which aggravate the Aβ pathology 24. On the other hand, activation of these cells can also arouse the clearance of Aβ plaques through phagocytosis 25, thus representing a double role for inflammation on amyloid pathology. In this review, we attempt to highlight the role of inflammation in AD.
Pathological hallmarks of AD
Currently, two pathological hallmarks are required in post-mortem observation of AD brain for the confirmation of AD diagnosis viz: 1) intracellular neurofibrillary tangles (NFTs), in which the main constituent is tau (hyperphosphorylated tau protein), and 2) extracellular insoluble senile plaques (SP) composed of amyloid beta protein 262728. An increased ratio of fibrillogenic Aβ42 was observed in most of the mutations that cause familial AD (FAD) 29. Recent discoveries have revealed that intracellular Aβ42 and soluble Aβ oligomers (2e6 peptides), which are present in neurons at the early stage of AD, exert more toxic effects than amyloid plaques 293031. Mutations of genes presenilin1 or presenelin2, which are responsible for encoding APP, can lead to FAD; these were observed in several mouse models of AD that have been developed 32.
Production of Aβ is carried out through an unusual pathway by proteolysis of APP. The APP is cut by beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) at position 99 of the C-terminus. Consequently, the C99-fragment is additionally cleaved by γ-secretase causing Aβ formation. On the other hand, in the usual pathway, the proteolysis of APP is done by α-secretases which cleave inside the Aβ precursor region, hence avoiding the formation of Aβ 29. The other pathological hallmark of AD is the presence of NFTs 10. NFTs are located inside the pyramidal neurons and are made up of tau and hyperphosphorylated tau proteins.
Under normal physiological conditions, tau is a soluble protein shouldered with the responsibility of maintaining the stability of microtubules and helps in the axonal transport. Under certain conditions these tau proteins become hyperphosphorylated, thus making them insoluble and as well as sticky, causing them to aggregate and form filaments which have no affinity to microtubules 33. Hence, the microtubules become destabilized. The destabilization of the microtubules affects the axonal transport of the cognitive neurotransmitters thereby affecting memory and learning ability.
The link between inflammation and AD
Inflammatory mechanisms in AD
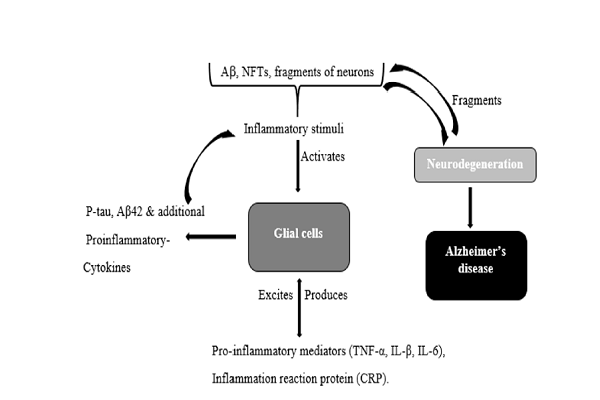
The inflammatory hypothesis is one of the prominent hypothesis put forward to describe the pathogenesis of AD. It is based on the tenet that “self-perpetuating progressive inflammation in the brain culminates in neurodegeneration” (Figure 1) 34. Presently, there is no any local inciting inflammatory factor that is associated with AD, although it is proposed that inflammation can be induced by the pathological hallmarks of AD (Aβ peptide plague and NFTs) or fragments of degenerated neurons 35. These pathological alterations are thought to excite glial cells to release pro-inflammatory cytokines (e.g. tumor necrosis factor alpha (TNF)-α, interleukin (IL)-1β, IL-6,) and inflammation reactive proteins (e.g. C-reactive protein (CRP)). The increased level of CRP and pro-inflammatory cytokines could then act either through autocrine or paracrine pathways, or both, to excite glial cells for additional production of p-tau, Aβ42, and pro-inflammatory molecules. Therefore, a positively self-supporting sequence is created in which inflammatory mediators perform a double role via stimulating glial cells and via activation of molecular pathway causing neurodegeneration, as reviewed by Angela Kamer et al. 36. This model is being supported by many pieces of evidence. Senile plaques are associated with activated microglia cells, reactive astrocytes, immunoreaction with antibodies (against C-reactive protein (CRP), TNF-α, IL-6, and IL-1), and complement proteins 35. TNF-α, IL-6, and IL-1 have the capability to induce synthesis of Aβ42 and phosphorylation of tau protein, while p-tau and Aβ42 can cause the production of TNF-α, IL-6 and IL-1 by glial cells 353738.

There are few clinical studies supporting the role of inflammation in the pathogenesis of AD. These studies explored the role played by systemic inflammatory markers and CRP in forecasting the starting point of AD. High level of CRP increased the chance of developing both cognitive decline 39 and AD in different populations 40. A study conducted in Honolulu evaluated 1,050 subjects and reported that an elevated level of CRP increased the likelihood of AD development 25 years later in life 41.
Pathogenesis of AD- Involvement of peripheral inflammatory mechanism
The existence of activated glial cell that can produce elevated levels of inflammatory molecules is one of the indicators of AD. When these inflammatory molecules are low, they might be protective. Nevertheless, at higher levels as in AD, they can stimulate neurodegeneration 42, which implies that any process capable of increasing the levels of inflammatory molecules contribute to the development and progression of AD. Peripheral pro-inflammatory molecules could also increase the brain inflammatory molecule pool in two mechanistic pathways, either via neuronal pathways or systemic circulation.
In the neuronal pathway, afferent fibers of the peripheral nerves are stimulated by peripheral cytokines which increase the expression of brain cytokines, or they can gain access into the brain compartments or channels associated with peripheral nerves. Once inside the brain, even if it is confined locally, they might increase the levels of brain cytokines significantly 43.
The systemic circulation pathway is an alternative mechanism through which pro-inflammatory molecules could get access into the central nervous system (CNS). They can enter via areas deficient of blood-brain barrier (BBB). Furthermore, they can enter through areas of the BBB by: (1) using cytokines specific transporters, (2) crossing through fenestrated capillaries in BBB, or (3) increasing the permeability of the BBB. Once in the brain, the pro-inflammatory molecules might indirectly stimulate the glial cells to give rise to more pro-inflammatory cytokines or they could directly upsurge the local pro-inflammatory cytokines. Once the glial cells are previously primed or activated as in AD, stimuli by pro-inflammatory cytokines coming from the periphery would result in overexpression of pro-inflammatory molecules, as reviewed by Kamer et al. 36.
Pathogenesis of AD- Peripheral infection
A study conducted on 135 healthy subjects and 128 AD patients provided evidence of the various kinds of infections and burdens originating from bacteria. These infectious agents included Helicobacter pylori, Chlamydophila pneumoniae, and Borrelia burgdorferi, and viruses (cytomegalovirus and herpes simplex virus type 1 are associated with AD) 15. A strong relationship was observed between high level of infectious burden and increased serum levels of Aβ markers and/or inflammatory cytokines in patients living with AD.
Chronic infections by these pathogens were shown to cause cardio-cerebral vascular disorders 44, which consequently encouraged the development of AD 45. That study provides a strong proof that cumulative infections are associated with AD and supports the role of infectious agents and inflammation in the etiopathogenesis of AD. Recently, several articles have demonstrated the involvement of infectious agents, such as fungus 46, protozoan 47, virus 48 and bacteria 49, in the etiology of AD. These agents were found to be linked to cognitive impairment and also implicated in the initiation and advancement of AD. In their review in 2014, Licastro et al. reported an association between chronic bacterial inflammation and Herpes family virus infections 48 in periodontal disease with AD. The study deliberated upon the probable pathogenesis of AD in association with chronic infections and pointed out the participation of age-dependent body immunity impairment, chronic activation of CNS, and systemic inflammatory responses in the development of AD 50.
Role of inflammation on tau pathology in AD
Induction of tau phosphorylation in tau models
The role of inflammation in aggravating the tau pathology was first seen from in vitro studies in primary microglia cells stimulated by lipopolysaccharide (LPS) or Aβ before being cultured with primary neocortical neurons 51. This revolutionary study disclosed that secretion of IL-1β (a pro-inflammatory cytokine) by microglial stimulation leads to elevation of tau phosphorylation through activation of p38-mitogen-activated protein kinases (MAPK). This was further reaffirmed in vivo using a 3xTg model that displayed both amyloid and tau pathologies 52. Chronic administration of LPS (0.5 mg/kg twice weekly for six weeks) activated tau phosphorylation at several phosphorylation sites associated with pre- and post- tau tangle pathology in 3xTg mice, as well as in early and advanced pathological stages 53. Furthermore, microglial activation and subsequent release of IL-1β were involved through stimulation of glycogen synthase kinase-3β (GSK-3β) 54 or cyclin-dependent kinase-5 (CDK-5) 54. Chronic overexpression of tumor necrosis factor-α (TNF-α), a pro-inflammatory cytokine, led to an increase in the pre-tangle-associated pT231 epitope 55.
On the other hand, when interferon-γ (IFNγ), another pro-inflammatory cytokine which is prominently linked to viral infections, was overexpressed, dephosphorylation of tau occured at the pre-tangle phosphorylation positions 56. Cumulatively, these observations suggested that the stimulation or inhibition of aggravation of tau pathology partly relies on the type of pro-inflammatory cytokines or immunological stressor present at the time. Nevertheless, there is evidence that chronic administration of NSAIDs (r-flurbiprofen or ibuprofen) causes tau dephosphorylation 57585960, in spite of poor penetration to the brain 61, which suggests that basal peripheral inflammation contributes to the progress of tau pathology, as reviewed by 20.
Alterations in tau pathogenesis
Tau phosphorylation plays a key role in triggering a pathological cascade that eventually culminates in the formation of NFT. There is a paucity of knowledge on the link associating the development of AD pathology to tau phosphorylation since the latter does not always develop into tau aggregates. For illustration, Sy et al. 54 reported an increased level of tau aggregation after chronic administration of LPS in the 3xTg model. On the contrary, Lee 62 reported an increase in tau phosphorylation with no effect on tau aggregation following acute LPS treatment in the rTG510 model. It is worth noting that the phosphorylation epitopes affected in both studies were the same 5462, perhaps demonstrating the role of chronic versus acute effects, even though amyloid might have played a role in the chronic study 63.
Recent development has shown that accumulation of tau oligomers at the synapses (instead of NFTs) makes them the most dangerous species with great pathogenic potential, e.g. they are believed to cause synaptic malfunction 64. This is believed to occur through a process called tau seeding, in which the tau oligomers have a special capability to propagate their pathology to healthy neighboring neurons by crossing through synapses 64. Evaluating the influence of inflammation on extracellular tau proteins will facilitate a superior understanding of the inflammatory process in tau pathology.

Relevance of inflammation in tau pathology of AD
It has been proposed that inflammation plays a significant part in tau pathology. However, the fundamental mechanistic pathways have remained vague. Although IL-1β signaling in microglia is linked to damage, the relevance of the tau phosphorylation in tau pathology is not well-understood. Pro-inflammatory cytokines significantly induce tau phosphorylation, but whether or not this continues to form tau aggregates is elusive. On the other hand, there are evidence which suggest a beneficial role of inflammation on tau pathology via induction of microglial phagocytosis of tau oligomers, with the possibility of inhibiting the spread of tau pathology. Based on the data from this review, inflammation is highly involved in tau pathology. However, most of the work on tau protein has come from preclinical studies, therefore, clinical studies on the role of inflammation in tau pathology is highly recommended.
Role of inflammation on Aβ pathology in AD
There are clear indications that inflammation occurs in pathologically vulnerable portions of the AD brain and is associated with NFT formation, Aβ deposit, and neurite damage in those areas. A multifaceted series of inflammatory mediators can be upregulated in response to inflammation within the brain 35 Figure 2. The cytopathology as a response from inflammation (complement activation, microgliosis, increased cytokine expression, astrocytosis, and acute phase protein response) is thought to be representative of secondary responses following early accumulation of Aβ in the brain 65. The immune response in the brain that follows Aβ deposition results in the accumulation of inflammatory mediators including free radicals, IL-1, IL-6, TNFα and microglia activation, as reviewed by Verdile et al. 66.
Even though inflammation seems to be a secondary response to Aβ accumulation, it is still very important in AD pathology, and there are abundant evidence that have established the role of inflammation in AD neurodegeneration. Firstly, the mechanisms of inflammation occur in the region of AD brain showing high AD pathology; inflammation occurs less frequently or is absent in regions with less AD pathology, such as the cerebellum 35 . Secondly, transgenic animals that exhibit inflammatory cytokines show profound pathological changes, including demyelination, neurodegeneration, and gliosis, as well as astrocytic and microglial activation 67. Thirdly, several clinical trials using anti-inflammatory drugs have reported that these drugs slow the progression or cause a delay in the onset of AD 35.
In AD development, production of Aβ and the failure of clearance of Aβ play very important roles. The overall impairment in the clearance of Aβ in the brain has been found in AD. Even though in the early stage, microglia can promote the clearance of Aβ and prevent the pathologic progression in AD. Meanwhile, a tenacious microglial buildup can also release cytotoxic molecules (e.g. reactive nitrogen species (RNS), ROS, chemokines, complement proteins and pro-inflammatory cytokines) which can now elevate the production of Aβ and reduce the rate of its clearance 6869 through the process of irregular function of astrocytes and microglia 7068, decreased expression of Aβ-degrading enzymes, and loss of structural and function integrity of BBB. Therefore, from these reports, inflammation can either increase or decrease AD pathology.
Conclusion
Inflammation has been found to be an integral part of Alzheimer’s disease. Some scientists believe that inflammation is the primary cause of AD, while others suggest that inflammation occurs because some proteins are deposited on the brain and the resulting inflammation aggravates the situation. On the other hand, other researchers see inflammation as an innate immunity that helps to defend the brain from neurotoxic proteins deposited on the brain. Whether inflammation causes AD or whether inflammation is a defensive mechanism in AD, it can be confirmed that inflammation is a fundamental part of AD. Further research should be conducted on inflammatory targeted drugs and the role of inflammation in AD.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License (CCBY4.0) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
List of abbreviations
AD: Alzheimer's disease; APP: Amyloid Precussor Protein; Aβ: Beta amyloid; BACE1: beta-site amyloid precursor protein cleaving enzyme 1; CDK-5: Cyclin dependent kinase-5; CRP: C-Reactive Protein; FAD: Familial Form of Alzheimer's Disease; GSK3β: Glucose synthase kinase 3 beta; GWAS: Genome-wide Association Studies; IFN-γ: Interferon-gamma; IL-1β: Interleukin 1beta; IL-6: Interleukin 6; MAPK: Mitogen activated protein kinase; NFTs: Neurofibrillary Tangles; NSAIDs: Non-steroidal anti-inflammatory Drugs; RNS: Reactive nitrogen species; ROS: Reactive oxygen species; SP: Senile Plaque; TNF-α: Tumor necrosis factor alpha
Competing interests
The authors declare that they have no conflicts of interest.
Funding
The authors would like to acknowledge Universiti Putra Malaysia for funding this research project (Grant number GP-IPS 9535400).
Authors' contributions
All authors contributed to the design of the research. MSC and SJ extracted the data and summarized it. MTBH, CNMT, ZA and MAMM edited the first draft. All authors reviewed, commented and approved the final draft.
References
-
Fischer
P.,
Zehetmayer
S.,
Jungwirth
S.,
Weissgram
S.,
Krampla
W.,
Hinterberger
M..
Risk factors for Alzheimer dementia in a community-based birth cohort at the age of 75 years. Dementia and Geriatric Cognitive Disorders.
2008;
25
:
501-7
.
-
Caselli
R. J.,
Beach
T. G.,
Yaari
R.,
Reiman
E. M..
Alzheimer’s disease a century later. The Journal of Clinical Psychiatry.
2006;
67
:
1784-800
.
-
Goedert
M.,
Spillantini
M. G.,
Cairns
N. J.,
Crowther
R. A..
Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron.
1992;
8
:
159-68
.
-
Goedert
M..
Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends in Neurosciences.
1993;
16
:
460-5
.
-
Finder
V. H..
Alzheimer’s disease: a general introduction and pathomechanism. Journal of Alzheimer\'s Disease.
2010;
22
:
5-19
.
-
Sisodia
S. S.,
Price
D. L..
Role of the beta-amyloid protein in Alzheimer’s disease. The FASEB Journal.
1995;
9
:
366-70
.
-
Koepsell
T. D.,
Kurland
B. F.,
Harel
O.,
Johnson
E. A.,
Zhou
X. H.,
Kukull
W. A..
Education, cognitive function, and severity of neuropathology in Alzheimer disease. Neurology.
2008;
70
:
1732-9
.
-
Roe
C. M.,
Xiong
C.,
Miller
J. P.,
Morris
J. C..
Education and Alzheimer disease without dementia: support for the cognitive reserve hypothesis. Neurology.
2007;
68
:
223-8
.
-
Placanica
L.,
Zhu
L.,
Li
Y. M..
Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS One.
2009;
4
:
e5088
.
-
Cummings
J.,
Lee
G.,
Mortsdorf
T.,
Ritter
A.,
Zhong
K..
Alzheimer’s disease drug development pipeline: 2017. Alzheimer\'s & Dementia: Translational Research & Clinical Interventions.
2017;
3
:
367-84
.
-
Alzheimer’s
Association.
Alzheimer’s disease facts and figures. Alzheimer\'s & Dementia.
2017;
13
:
325-73
.
-
Association
A..
Alzheimer’s disease facts and figures. Alzheimer\'s & Dementia.
2018;
14
:
367-429
.
-
Serrano-Pozo
A.,
Mielke
M. L.,
Gómez-Isla
T.,
Betensky
R. A.,
Growdon
J. H.,
Frosch
M. P..
Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. American Journal of Pathology.
2011;
179
:
1373-84
.
-
Lambert
J. C.,
Ibrahim-Verbaas
C. A.,
Harold
D.,
Naj
A. C.,
Sims
R.,
Bellenguez
C.,
European Alzheimer’s Disease
Initiative,
Genetic
Environmental Risk in Alzheimer’s
Disease,
Alzheimer’s Disease Genetic
Consortium,
Cohorts for
Heart,
Aging Research in Genomic
Epidemiology.
Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genetics.
2013;
45
:
1452-8
.
-
Bu
X. L.,
Yao
X. Q.,
Jiao
S. S.,
Zeng
F.,
Liu
Y. H.,
Xiang
Y..
A study on the association between infectious burden and Alzheimer’s disease. European Journal of Neurology.
2015;
22
:
1519-25
.
-
Budni
J.,
Garcez
M. L.,
de Medeiros
J.,
Cassaro
E.,
Bellettini-Santos
T.,
Mina
F..
The Anti-Inflammatory Role of Minocycline in Alzheimer´s Disease. Current Alzheimer Research.
2016;
13
:
1319-29
.
-
Zilka
N.,
Kazmerova
Z.,
Jadhav
S.,
Neradil
P.,
Madari
A.,
Obetkova
D..
Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. Journal of Neuroinflammation.
2012;
9
:
47
.
-
Tejera
D.,
Heneka
M. T..
Microglia in Alzheimer’s disease: the good, the bad and the ugly. Current Alzheimer Research.
2016;
13
:
370-80
.
-
Jaturapatporn
D.,
Isaac
M. G.,
McCleery
J.,
Tabet
N..
Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database of Systematic Reviews.
2012;
:
CD006378
.
-
Barron
M.,
Gartlon
J.,
Dawson
L. A.,
Atkinson
P. J.,
Pardon
M. C..
A state of delirium: deciphering the effect of inflammation on tau pathology in Alzheimer’s disease. Experimental Gerontology.
2017;
94
:
103-7
.
-
Wang
J.,
Tan
L.,
Wang
H. F.,
Tan
C. C.,
Meng
X. F.,
Wang
C..
Anti-inflammatory drugs and risk of Alzheimer’s disease: an updated systematic review and meta-analysis. Journal of Alzheimer\'s Disease.
2015;
44
:
385-96
.
-
Michaud
J. P.,
Rivest
S..
Anti-inflammatory signaling in microglia exacerbates Alzheimer’s disease-related pathology. Neuron.
2015;
85
:
450-2
.
-
Andreasson
K. I.,
Bachstetter
A. D.,
Colonna
M.,
Ginhoux
F.,
Holmes
C.,
Lamb
B..
Targeting innate immunity for neurodegenerative disorders of the central nervous system. Journal of Neurochemistry.
2016;
138
:
653-93
.
-
Brugg
B.,
Dubreuil
Y. L.,
Huber
G.,
Wollman
E. E.,
Delhaye-Bouchaud
N.,
Mariani
J..
Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proceedings of the National Academy of Sciences of the United States of America.
1995;
92
:
3032-5
.
-
Fiala
M.,
Cribbs
D. H.,
Rosenthal
M.,
Bernard
G..
Phagocytosis of amyloid-beta and inflammation: two faces of innate immunity in Alzheimer’s disease. Journal of Alzheimer\'s Disease.
2007;
11
:
457-63
.
-
Querfurth
H. W.,
LaFerla
F. M..
Alzheimer’s disease. The New England Journal of Medicine.
2010;
362
:
329-44
.
-
Bulk
M.,
Kenkhuis
B.,
van der Graaf
L. M.,
Goeman
J. J.,
Natte
R.,
van der Weerd
L..
Post-mortem T2*-weighted MRI imaging of cortical iron reflects severity of Alzheimer’s Disease. bioRxiv.
2018;
31
:
1-19
.
-
Edler
M. K.,
Sherwood
C. C.,
Meindl
R. S.,
Hopkins
W. D.,
Ely
J. J.,
Erwin
J. M..
Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease. Neurobiology of Aging.
2017;
59
:
107-20
.
-
LaFerla
F. M.,
Green
K. N.,
Oddo
S..
Intracellular amyloid-beta in Alzheimer’s disease. Nature Reviews. Neuroscience.
2007;
8
:
499-509
.
-
Cimini
S.,
Sclip
A.,
Mancini
S.,
Colombo
L.,
Messa
M.,
Cagnotto
A..
The cell-permeable Aβ1-6A2VTAT(D) peptide reverts synaptopathy induced by Aβ1-42wt. Neurobiology of Disease.
2016;
89
:
101-11
.
-
Castillo
W. O.,
Aristizabal-Pachon
A. F..
Galantamine protects against beta amyloid peptide-induced DNA damage in a model for Alzheimer’s disease. Neural Regeneration Research.
2017;
12
:
916-7
.
-
Kitazawa
M.,
Medeiros
R.,
Laferla
F. M..
Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Current Pharmaceutical Design.
2012;
18
:
1131-47
.
-
Alonso
A.,
Zaidi
T.,
Novak
M.,
Grundke-Iqbal
I.,
Iqbal
K..
Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proceedings of the National Academy of Sciences of the United States of America.
2001;
98
:
6923-8
.
-
McGeer
P. L.,
McGeer
E. G..
Inflammation, autotoxicity and Alzheimer disease. Neurobiology of Aging.
2001;
22
:
799-809
.
-
Akiyama
H.,
Barger
S.,
Barnum
S.,
Bradt
B.,
Bauer
J.,
Cole
G. M..
Inflammation and Alzheimer’s disease. Neurobiology of Aging.
2000;
21
:
383-421
.
-
Kamer
A. R.,
Craig
R. G.,
Dasanayake
A. P.,
Brys
M.,
Glodzik-Sobanska
L.,
de Leon
M. J..
Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimer\'s & Dementia.
2008;
4
:
242-50
.
-
Konsman
J. P.,
Drukarch
B.,
Van Dam
A. M..
(Peri)vascular production and action of pro-inflammatory cytokines in brain pathology. Clinical Science (London, England).
2007;
112
:
1-25
.
-
Gosselin
D.,
Rivest
S..
Role of IL-1 and TNF in the brain: twenty years of progress on a Dr. Jekyll/Mr. Hyde duality of the innate immune system. Brain, Behavior, and Immunity.
2007;
21
:
281-9
.
-
Yaffe
K.,
Kanaya
A.,
Lindquist
K.,
Simonsick
E. M.,
Harris
T.,
Shorr
R. I..
The metabolic syndrome, inflammation, and risk of cognitive decline. Journal of the American Medical Association.
2004;
292
:
2237-42
.
-
Engelhart
M. J.,
Geerlings
M. I.,
Meijer
J.,
Kiliaan
A.,
Ruitenberg
A.,
van Swieten
J. C..
Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Archives of Neurology.
2004;
61
:
668-72
.
-
Schmidt
R.,
Schmidt
H.,
Curb
J. D.,
Masaki
K.,
White
L. R.,
Launer
L. J..
Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia Aging Study. Annals of Neurology.
2002;
52
:
168-74
.
-
von Bernhardi
R.,
Eugenín
J..
Microglial reactivity to β-amyloid is modulated by astrocytes and proinflammatory factors. Brain Research.
2004;
1025
:
186-93
.
-
Miller
AJ,
Luheshi
GN,
Rothwell
NJ,
Hopkins
SJ.
Local cytokine induction by LPS in the rat air pouch and its relationship to the febrile response. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology.
1997;
272
:
R857-R861
.
-
Strandberg
T. E.,
Pitkala
K. H.,
Linnavuori
K. H.,
Tilvis
R. S..
Impact of viral and bacterial burden on cognitive impairment in elderly persons with cardiovascular diseases. Stroke.
2003;
34
:
2126-31
.
-
Li
J.,
Wang
Y. J.,
Zhang
M.,
Xu
Z. Q.,
Gao
C. Y.,
Fang
C. Q.,
Chongqing Ageing Study
Group.
Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology.
2011;
76
:
1485-91
.
-
Pisa
D.,
Alonso
R.,
Juarranz
A.,
Rábano
A.,
Carrasco
L..
Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. Journal of Alzheimer\'s Disease.
2015;
43
:
613-24
.
-
Prandota
J..
Possible link between Toxoplasma gondii and the anosmia associated with neurodegenerative diseases. American Journal of Alzheimer\'s Disease and Other Dementias.
2014;
29
:
205-14
.
-
Licastro
F.,
Carbone
I.,
Raschi
E.,
Porcellini
E..
The 21st century epidemic: infections as inductors of neuro-degeneration associated with Alzheimer’s Disease. Immunity & Ageing.
2014;
11
:
22
.
-
Maheshwari
P.,
Eslick
G. D..
Bacterial infection and Alzheimer’s disease: a meta-analysis. Journal of Alzheimer\'s Disease.
2015;
43
:
957-66
.
-
Lim
S. L.,
Rodriguez-Ortiz
C. J.,
Kitazawa
M..
Infection, systemic inflammation, and Alzheimer’s disease. Microbes and Infection.
2015;
17
:
549-56
.
-
Li
Y.,
Liu
L.,
Barger
S. W.,
Griffin
W. S..
Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience.
2003;
23
:
1605-11
.
-
Oddo
S.,
Caccamo
A.,
Shepherd
J. D.,
Murphy
M. P.,
Golde
T. E.,
Kayed
R..
Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron.
2003;
39
:
409-21
.
-
Kitazawa
M.,
Oddo
S.,
Yamasaki
T. R.,
Green
K. N.,
LaFerla
F. M..
Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience.
2005;
25
:
8843-53
.
-
Sy
M.,
Kitazawa
M.,
Medeiros
R.,
Whitman
L.,
Cheng
D.,
Lane
T. E..
Inflammation induced by infection potentiates tau pathological features in transgenic mice. American Journal of Pathology.
2011;
178
:
2811-22
.
-
Janelsins
M. C.,
Mastrangelo
M. A.,
Park
K. M.,
Sudol
K. L.,
Narrow
W. C.,
Oddo
S..
Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. American Journal of Pathology.
2008;
173
:
1768-82
.
-
Mastrangelo
M. A.,
Sudol
K. L.,
Narrow
W. C.,
Bowers
W. J..
Interferon-{γ} differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. American Journal of Pathology.
2009;
175
:
2076-88
.
-
McKee
A. C.,
Carreras
I.,
Hossain
L.,
Ryu
H.,
Klein
W. L.,
Oddo
S..
Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Research.
2008;
1207
:
225-36
.
-
Carreras
I.,
McKee
A. C.,
Choi
J. K.,
Aytan
N.,
Kowall
N. W.,
Jenkins
B. G..
R-flurbiprofen improves tau, but not Aß pathology in a triple transgenic model of Alzheimer’s disease. Brain Research.
2013;
1541
:
115-27
.
-
Šimić
G.,
Babić Leko
M.,
Wray
S.,
Harrington
C.,
Delalle
I.,
Jovanov-Milošević
N..
Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules.
2016;
6
:
6
.
-
Héraud
C.,
Goufak
D.,
Ando
K.,
Leroy
K.,
Suain
V.,
Yilmaz
Z..
Increased misfolding and truncation of tau in APP/PS1/tau transgenic mice compared to mutant tau mice. Neurobiology of Disease.
2014;
62
:
100-12
.
-
Parepally
J. M.,
Mandula
H.,
Smith
Q. R..
Brain uptake of nonsteroidal anti-inflammatory drugs: ibuprofen, flurbiprofen, and indomethacin. Pharmaceutical Research.
2006;
23
:
873-81
.
-
Lee
D. C.,
Rizer
J.,
Selenica
M. L.,
Reid
P.,
Kraft
C.,
Johnson
A..
LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. Journal of Neuroinflammation.
2010;
7
:
56
.
-
Noble
W.,
Garwood
C.,
Stephenson
J.,
Kinsey
A. M.,
Hanger
D. P.,
Anderton
B. H..
Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. The FASEB Journal.
2009;
23
:
739-50
.
-
Guerrero-Muñoz
M. J.,
Gerson
J.,
Castillo-Carranza
D. L..
Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Frontiers in Cellular Neuroscience.
2015;
9
:
464
.
-
Weiner
H. L.,
Selkoe
D. J..
Inflammation and therapeutic vaccination in CNS diseases. Nature.
2002;
420
:
879-84
.
-
Verdile
G.,
Fuller
S.,
Atwood
C. S.,
Laws
S. M.,
Gandy
S. E.,
Martins
R. N..
The role of beta amyloid in Alzheimer’s disease: still a cause of everything or the only one who got caught?. Pharmacological Research.
2004;
50
:
397-409
.
-
Wyss-Coray
T.,
Masliah
E.,
Mallory
M.,
McConlogue
L.,
Johnson-Wood
K.,
Lin
C..
Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature.
1997;
389
:
603-6
.
-
Brandenburg
L. O.,
Konrad
M.,
Wruck
C. J.,
Koch
T.,
Lucius
R.,
Pufe
T..
Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1-42-induced signal transduction in glial cells. Journal of Neurochemistry.
2010;
113
:
749-60
.
-
Feng
Y.,
Li
L.,
Sun
X. H..
Monocytes and Alzheimer’s disease. Neuroscience Bulletin.
2011;
27
:
115-22
.
-
Yang
C. N.,
Shiao
Y. J.,
Shie
F. S.,
Guo
B. S.,
Chen
P. H.,
Cho
C. Y..
Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiology of Disease.
2011;
42
:
221-30
.
Comments
Downloads
Article Details
Volume & Issue : Vol 5 No 8 (2018)
Page No.: 2552-2564
Published on: 2018-08-21
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 10998 times
- Download PDF downloaded - 2534 times
- View Article downloaded - 0 times
