

Copyrights: Nguyen Thi Huynh Nga, Bui Chi Bao, Nguyen Vuong Thao Vy, Nguyen Manh Cong, Nguyen Minh Hiep, 2022. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Introduction: Whole-exome sequencing (WES), a high-throughput DNA sequencing method, has swiftly gained traction as a scalable approach for detecting novel de novo mutations and generating molecular diagnoses for genetic illnesses.
Case presentation: We describe a neonate girl with a novel de novo desmin (DES) variant. The patient was referred to our Department of Cardiology right after birth for multiple clinical cardiac and pulmonary presentations. Her family history was negative for cardiovascular diseases while her echocardiographic diagnosis indicated macrocardia, arrhythmia, ventricular septal defect (VSD) in association with tricuspid regurgitation. Electrocardiography showed prolonged QTc interval, premature atrial contractions following pulmonary arterial hypertension. The patient had a surgical VSD closure at the age of seven months. The healthy infant was fit enough to be discharged six weeks later. Whole-exome sequencing identified a novel de novo heterozygous frameshift deletion mutation p.E246VfsTer4 in DES gene. Among the variants detected, mutation in DES was predicted to be the only pathogenic one, causing desminopathy phenotype for the patient.
Conclusion: This report strengthens the genetic aetiology for desminopathy and highlights the need for screening an intriguing candidate, those who have the DES gene in congenital cardiovascular probands.
Introduction
Desminopathy refers to the genetically and phenotypically heterogeneous group of myofibrillar myopathies (MFM) caused mainly by desmin gene (DES) mutations1, 2. Mutations of DES causes progressive myopathies, cardiomyopathies, cardiac conduction defects, arrhythmias, pulmonary and miscellaneous disease manifestations3, 4, 5. Desminopathy has a complicated aetiology, including both hereditary or sporadic myopathies causes1. In addition, the mode of inheritance can be AD or AR1, 6. Currently, mutations have been identified in every single domain of DES1, 7. Even though mutations are identical or located adjacent to one other, DES mutations are pleiotropic, creating numerous distinct phenotypes6, 7. For cardiomyopathies, DES mutations can lead to dilated cardiomyopathy (DCM)8, hypertrophic cardiomyopathy (HCM)9, restrictive cardiomyopathy (RCM)10, arrhythmogenic cardiomyopathy (ACM)11 or left ventricular noncompaction cardiomyopathy (LVNC)12. Because DES mutations induce clinically symptomatic and asymptomatic ECG abnormalities in cardiac illnesses, the genotype-phenotype relationship is complicated, making patient management problematic1. Moreover, different DES mutations lead to multiple phenotypes within the same families6. Gene dosage effects caused by heterozygous, homozygous, double heterozygous and compound heterozygous mutations are also considered6. However, in some cases, heterozygous family members did not develop a phenotype excluding haploinsufficiency as the main molecular mechanism13, 14, 15. In brief, the complexity of genetics, epigenetics and phenotypic variability among the desminopathies limit the ability to establish a diagnosis based solely on clinical features. Accordingly, specific diagnoses and prognoses for desminopathy patients greatly rely on novel and powerful high throughput technologies and genome-wide association studies.
Whole-exome sequencing (WES), a high-throughput DNA sequencing technology, has quickly become a scalable tool, generating a molecular diagnosis for Mendelian genetic disorders16, 17, 18. Exome, a collection of all exons within the genome, comprises approximately 1% of the total genome and harbours 85% of interpretable mutations that result in clinical phenotypes19, 20. This 1% coding region has a less repetitive sequence than the noncoding ones. Identifying and sequencing of the exome is computationally and analytically less intense, therefore16, 17. WES has become a powerful tool in identification of the causal variant(s) for a disease of interest18.
This study herein reports on the family of a newborn girl with multiple clinical cardiac and pulmonary presentations diagnosed with desminopathy as the patient carries a frameshift mutation p.E246VfsTer4 in DES gene. The report strengthens the genetic aetiology for desminopathy and highlights the need of screening an intriguing candidate, the DES gene in congenital cardiovascular probands.
Case presentation
Material and methods
Library preparation and whole-exome sequencing
Genomic DNA was isolated from white blood cells using standard techniques (DNA Extraction Kit, Qiagen, Hilden, Germany). Adaptors and two paired-end DNA libraries with insert sizes ranging from 100 to 900 bp were used to ligate DNA fragments. After polymerase chain reaction (PCR) enrichment, DNA libraries were produced for cardiac gene enrichment re-sequencing on a NovaSeq 6000 Sequencing System (Illumina, San Diego, CA, USA). The patient was screened for variants in 142 cardiomyopathy associated genes using the TruSeq™ Cardiomyopathy gene panel, resulting in the 150-bp paired-end sequencing reads with at least 100-fold average sequencing depth for each sample.
Alignment and mapping
Low quality reads were discarded from raw data to generate clean reads. Clean reads were aligned to the reference human genome UCSC Genome Build hg19. For reads alignment and base calling, Illumina automated workflow was used: Burrows–Wheeler Aligner (BWA), Genome Analysis Toolkit (GATK) and Sequence Alignment Map (SAM tools).
Variant annotation and filtering
For variant annotation, we used ANNOVAR and VEP software programs. Variants were then compared computationally with the list of reported pathogenic variations from the Human Gene Mutation Database (HGMD, professional version). We filtered extremely rare variants located in exon- and consensus splice-sequences (± 1 and 2 position) with a global minor allele frequency (MAF) ≤ 0.0005. Variants that are not in the HGMD, synonymous variants and intronic variants that were more than 15 bp from exon boundaries were not considered.
Variant classification
We used the guidelines of the American College of Medical Genetics and Genomics (ACMG) for variant classification to assess dominant single gene mutation with clear clinical impact on CMs (cardiomyopathies). The Exome Aggregation Consortium (ExAC) browser was used as a reference for allele frequencies. A variety of prediction techniques were used to evaluate the missense variant, including ENTPRISE-X, MutationTaster, PROVEAN, and SIFT. Multiple ortholog alignment with Alamut software was used to determine the degree of conservation of the damaged residue. (Interactive Biosoftware, Rouen, France).
Sanger sequencing
All variants of interest were confirmed with Sanger sequencing. Primers of the identified variants were designed using primer3. PCR primer pair used to amplify products was DES-F: 5’-TGGATGCAGCTACTCTAGCT-3’, DES-R: 5’-CAACTCACGGATCTCCTGTA-3’. Direct sequencing of the PCR products was performed on an ABI3130XL sequencer (Applied Biosystems) according to the manufacturer’s protocol.

Results
Index patient clinical information
A newborn baby girl with cardiomyopathy and arrhythmia is the subject of this research. The baby, who was born to healthy parents, is the second child of the family. Her family history was negative for cardiovascular diseases (CVDs), and both parents as well as her sibling were asymptomatic with arrhythmias, expressing normal ECGs (data not shown). The clinical status was indicated by the patient family pedigree (Figure 1A).
The foetal echocardiography at 26 weeks detected signs of congenital heart disease (CHD) during the prenatal period including: macrocardia and valvular heart disease — showing tricuspid valve (TV) did not close completely when the right ventricle contracted — diagnosed with grade 2/4.
Through echocardiography, the foetal heart at 32 weeks showed cardiac defects including: arrhythmia, cardiomegaly and ventricular septal defect (VSD) in association with tricuspid regurgitation (TR).
On February 1, 2019, at 39 weeks of gestation, the mother suffered from pre-eclampsia. A girl baby weighing 3,000 grams was delivered by emergency caesarean section in good condition.
The neonate's diagnosis at the time of hospital admission shows that the patient's haemodynamically stable state and her sinus rhythm was present at a regular rate of 120 beats per minute (bpm).
During hospitalization, the baby girl was well breastfed and gained weight gradually. By the ECG Holter monitor, the heart rate had been identified to be in a range between 130 and 160 bpm. However, electrocardiography showed a prolonged QTc interval ranging from 470 to 520 ms. Tall P wave (Figure 1B) showed arrythmias. Premature atrial contractions (PACs) occurred occasionally. Moreover, echocardiographies showed a 4 mm midmuscular VSD with a left-to-right shunt and TR grade 2/4. Pulmonary arterial hypertension (PAH) was also put into the clinical record as the pulmonary artery pressures (PAPs) reached 60 mmHg. Digital X-ray image showed cardiac enlargement (Figure 1C). Based on these clinical examinations, the patient was closely watched on cardiac shunt and TR progressions.
Four weeks later, on February 27, 2019, the infant girl reached 3,200 grams. Echocardiographies identified a 4 mm perimembranous VSD. The PAPs reached 65 mmHg. ECG recordings indicated a heart rate of around 150 bpm and a prolonged QTc interval of 450 ms. As a result, cardiologists had established a strong suspicion for LQTS type 2. The patient was then medicated with Viagra daily.
On May 14, 2019, the baby was still well breastfed and reached 4,800 grams. The ECG Holter monitor identified a heart rate of around 140 bpm and a prolonged QTc interval up to 480 ms. Echocardiographies showed a 5 mm VSD. The atrial septal defect (ASD) size was 3.5 mm. The PAPs reached 70 mmHg. Based on these clinical results, the patient was continually administrated with Viagra and a surgical VSD closure was indicated in July 2019.
On August 30, 2019, six weeks after the surgical VSD closure, echocardiograms revealed a left ventricular diameter of 26mm, PAPs of 50 mmHg, and cardiac ejection fraction (EF) ranging from 51 to 60%. The healthy infant weighed 6,500 grammes and was deemed suitable to be discharged.
| Item | Result |
|---|---|
| Total number of reads | 49,878,538 |
| Average read length (bp) Total yield (Mbp) Total read base (bp) GC content (%) Q20 (%) Q30 (%) | 151.0 7,532 7,531,659,238 49.37 97.71 93.73 |
| No | Gene | Chr | Exonic function | HGVS.c | HGVS.p |
|---|---|---|---|---|---|
| 1 | ABL1 | 9 | Synonymous SNV | c.C2787T | p.G929G |
| 2 | AHCY | 20 | Synonymous SNV | c.C393T | p.D131D |
| 3 | APOB | 2 | Nonsynonymous SNV | c.C1594T | p.R532W |
| 4 | CDK13 | 7 | Nonsynonymous SNV | c.C1207T | p.L403F |
| 5 | DES | 2 | Frameshift insertion | c.733_734insAGGT | p.E246VfsTer4 |
| 6 | PSEN2 | 1 | Synonymous SNV | c.C315T | p.T105T |
| 7 | SDHA | 5 | Nonsynonymous SNV | c.A1106G | p.Y369C |

Identification of a novel heterozygous desmin-gene mutation
To prevent patients who have heart failure or arrhythmias from sudden cardiac death (SCD), genetic testing could help to increase the diagnostic accuracy as well as facilitate clinical assessment and appropriate therapy of the patient. We therefore performed WES of the newborn girl who was diagnosed with CVDs to identify genetic variants of Vietnamese CM patients.
The genetic data of the subject are shown in Table 1. The Illumina sequencer yielded 49,878,538 reads with average 151 base pairs (bp) in length and the GC content was 49.37%.
To further analyse the mutations and identify potential genes, we narrowed down the potential candidates including 142 genes linked to cardiomyopathy (CM). These genes were further filtered based on the mapping quality MQ ≥ 40, prediction of protein alteration, missense variants and absence from SNP database of ADS. As a result, 7 variants in different genes have been identified (Table 2).
Our results showed that WES identified a novel heterozygous frameshift insertion variant in the desmin (DES) gene. The variant c.733_734insAGGT belonged near the end of exon 3, where a group of four nucleotides insertion occurred: AGGT was added at the position between 733 and 734 nucleotides in the cDNA, leading to the replacement of glutamic acid (Glu, E) by valine (Val, V) at the position 246 on the DES protein. The E246 was the first amino acid changed, the length of the shift frame is 4, including the termination codon (Figure 2). p.E246V produces a substitution of an acidic amino acid by a hydrophobic one. The physico-chemical properties between Glu and Val are much different.
In this particular case, the only potentially pathogenic variant found in our patient was p.E246VfsTer4 in DES (Table 2). p.E246VfsTer4 is not listed in public databases nor has it been identified in genotyping projects of general population comprising several thousands of subjects (Exome Variant Server, Exome Aggregation Consortium). Pathogenicity (variant pathogenic effect) was rated based on the presence of a previously associated or candidate gene. Given its extremely low MAF, we conclude that p.E246VfsTer4 is a mutation.
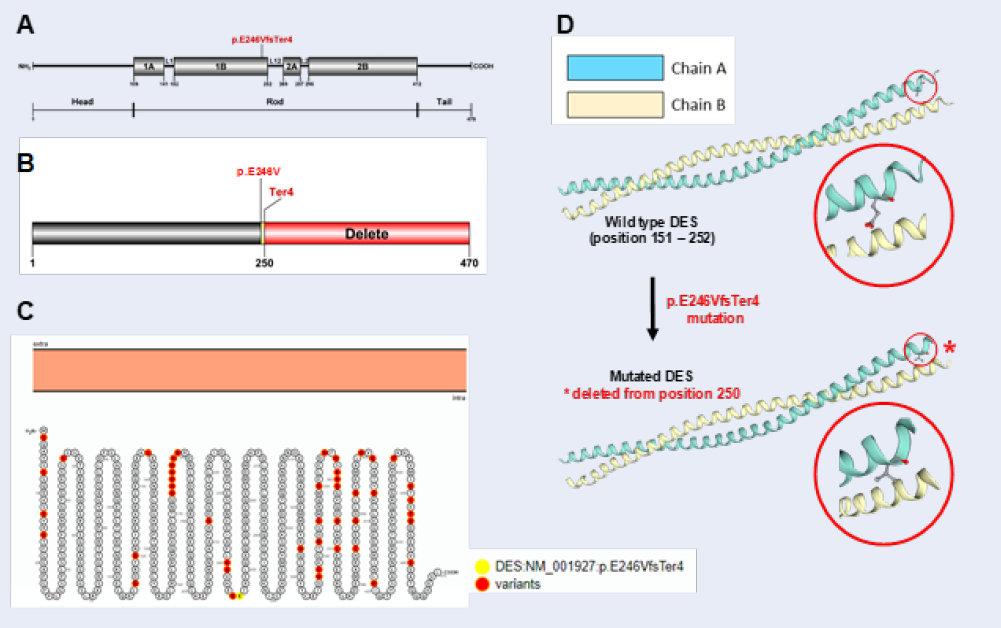
Figure 2A indicated that human mature DES is a 470 amino acid protein which comprises three major domains: a variable non- α-helical amino-terminal head, a conserved α-helical rod, and a non- α-helical carboxy-terminal tail21. The long central rod is interrupted by 3 short non-helical linkers (L1, L12 and L2), thus generating 4 helices (1A, 1B, 2A and 2B)6. Our novel p.E246VfsTer4 mutation is located at the 1B helical domain. Hence, the rest of the polypeptide chain was deleted (Figure 2B).

We also figure out the in silico predicted impact over the calmodulin protein, the 2015 American College of Medical Genetics and Genomics criteria as well as widely used software scores were considered. Five protein function prediction bioinformatics tools chosen were ENTPRISE-X, MutationTaster, Provean, SIFT and VarSome. All of this software classified this variant as likely pathogenic. Taken together, there is a very high possibility that this mutation is pathogenic and disease-causing in the patient.
To insist the possibility of the mutation p.E246VfsTer4 causing CVDs in the proband, WES was also performed in blood samples of the healthy parents (data not shown). The parents were both asymptomatic with no phenotypic abnormalities on the baseline or stress test ECGs. Additionally, they both had a normal resting heart rate in sinus rhythm and normal exercise-induced tachycardia (data not shown). As our hypothesis, the mutation was not present in the patient's parents, suggesting that the mutation p.E246VfsTer4 in DES was the cause of CVDs in the infant and a de novo one. Consistent with the high throughput sequencing, Sanger sequencing results of the trio revealed that the patient carried a frameshift mutation (Figure 3C) while her parents did not (Figure 3D-E).
Last but not least, DES phylogeny represents evolutionary relationships among eukaryotes (Figure 3F). In addition, the novel mutation p.E246VfsTer4 affected the highly conserved polypeptide region (Figure 3G), predicting a high pathogenic effect. Indeed, this mutation started at a naturally conservative glutamic acid at position 246, which is not changed when compared the protein sequence encoded by DES gene of human to those of other species (Figure 3G).
Discussion
In this study we aimed to discover the genetic basis for CVDs with overlapping features of other illnesses. On a newborn index patient with CM, we tested the efficiency of a new comprehensive next-generation sequencing CM gene panel assay and found that it discovered mutations with excellent sensitivity and precision. In complicated situations, the method helped establish a diagnosis and altered diagnoses based only on clinical features. The results showed that WES identified a novel heterozygous frameshift deletion in DES. This gene is located in a CM susceptibility region (chr2q35) and encodes muscle-specific protein DES, the main component of intermediate filaments that exert central role of myofibrils, the positioning of cell organelles and signaling events6, 7. Desmin mutations are the most frequently encountered gene defects in MFM1. Especially, various form of cardiological signs presence combinationally in majority of DES mutation carriers4. The morphological hallmarks of desmin cardiomyopathies are DES-positive protein aggregates, which can be found in subsarcolemmal, intermyofibrillar and perinuclear regions, leading to mitochondrial abnormalities, mis-shaped myonuclei, myocyte hypertrophy, disarray of the myocytes, focally lysed myofibrils and cytoplasmic vacuolar degeneration1, 22, 23. Consequently, DES mutations cause multiple pathomechanisms leading to death of cardiomyocytes and contributing accordingly to disease progression7. Particularly, mutations in the 1B segment of DES lead to the disruption of the filamentous network, resulting in severe clinical presentations. A 9-year-old Caucasian child, whose family history was negative for neuromuscular or cardiac disorders, suffered from SCD after being in a persistent vegetative state for 8 years. The 40-year-old father carried a heterozygous single-nucleotide insertion mutation occurring at codon 241 causes a frame shift leading to serial amino acid replacements: V242E, H243S, E244A and eventually a premature termination signal at codon 245. This patient presented with distal myopathy, arrhythmia and malignant ventricular tachycardia because the mutation induces collapse of a pre-existing desmin cytoskeleton, alters the subcellular distribution of mitochondria and triggers mitochondrial dysfunction through inhibition of complex I activity24. A heterozygous E245D missense mutation was found in a 9-year-old French girl with palpitations and right bundle branch block (RBBB). Her 33-year-old mother, who had the same mutation, was diagnosed with skeletal muscle involvement, third-degree atrioventricular block (AVB), paced arrhythmias, and hypertrophic CM, which necessitated pacemaker insertion25. A 52-year-old male with distal muscle weakness in his legs and arms, as well as distal muscle atrophy and cardiac arrhythmia, had the identical mutation E245D.His 42-year-old brother developed restrictive CM at the age of 20, required cardiac transplantation. Their cardiac and skeletal muscle biopsy specimens revealed granulofilamentous material and plaques containing numerous proteins, foremost DES26.
Advances in WES showed that approximately 10% of sporadic severe cases of CCDs are caused by de novo mutations. Multiple studies suggested that de novo mutations in DES lead to severe cardiac arrests in childhood1, 7. Age of disease onset and rate of progression depend on the type of inheritance and mutation site27. Compound heterozygosity for two autosomal recessive mutations A360P and N393I was detected in three out of four siblings characterized by childhood-onset aggressive course of cardiac and skeletal myopathy while neither of these two mutations was pathogenic in other family members. The children developed complete atrioventricular conduction block requiring a permanent pacemaker at 2, 9 and 10 years of age. The three siblings showed progressive muscle weakness and atrophy with swallowing and breathing difficulties in their early twenties. Several years later, the two older siblings died of progressive fibrosis of the cardiac conduction system and cardiomyopathy. The surviving one developed congestive heart failure secondary to restrictive cardiomyopathy. Their tissue examinations showed intracytoplasmic accumulation of amorphous DES distributed subsarcolemmally28. In another family with A120D mutation, three out of four siblings died from SCD as teenagers. The alive 34-year-old sister presented with atrial flutter, dilated atria and variable atrioventricular conduction caused by some polymorphic premature ventricular contractions (PVCs). The patient received an implantable cardioverter defibrillator. Her three cousins, all aged 13, also perished suddenly. The lack of DES staining within the intercalated disc and significant cytoplasmic aggregation development were discovered in ventricular tissue slices29.
The genetic test results of these young patients were suggestive of a hidden molecular diagnosis of desminopathies. In addition, the correlation between the patient’s symptoms and the known involvements of DES in the heart shows the effect of the mutation on protein function24, 25, 26, 27, 28, 29. DES is predicted to have a high pathogenic effect in CVDs since it is enriched at the level of intercalated discs in cardiac muscle and acts as a basis for a mechano-chemical signalling between various compartments to maintain a highly organized dynamic muscle network1.
Our findings strengthen the genetic aetiology for desminopathies as well as CVDs during infancy. Given that the 1B helical domain clusters various desminopathy-causing mutations (including frameshift, splice site, deletion and missense ones) which express variety of different cardiomyopathies with arrhythmias or conduction disorders, the functional importance of the specific topological domain seems to be intolerant to genetic variation1, 2, 6, 30. Therefore, as an intriguing candidate, DES gene must be screened in congenital CVD probands because a timely patient management may prevent disease progressions.
Conclusions
Our study herein reports on the family of a newborn girl with multiple clinical cardiac and pulmonary presentations diagnosed with desminopathy as the patient carries a frameshift mutation p.E246VfsTer4 in DES gene. The report strengthens the genetic aetiology for desminopathy and highlights the need of screening an intriguing candidate, the DES gene in congenital cardiovascular probands.
Abbreviations
CVD: cardiovascular disease, NGS: next-generation sequencing, SCD: sudden cardiac death, WES: whole exome sequencing
Acknowledgments
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106-YS.01-2016.39. We would also like to thank Truong Nhat Vi and Nguyen Minh Tri Viet for the contribution in this work.
Author’s contributions
Nguyen Thi Huynh Nga and Nguyen Minh Hiep wrote the manuscript. Bui Chi Bao, Nguyen Thi Huynh Nga and Nguyen Minh Hiep designed the study, analyzed and interpreted the data. Bui Chi Bao, Nguyen Vuong Thao Vy and Nguyen Manh Cong performed the experiments. All authors approved the final manuscript.
Funding
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106-YS.01-2016.39.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Ethics approval and consent to participate
Procedures were reviewed and approved by the Ethical Committee of the University of Medicine and Pharmacy, Ho Chi Minh, Viet Nam. Participant’s parents were consenting to including the research methods involved.
Consent for publication
The work described has not been published before, it is not under consideration for publication anywhere else, its publication has been approved by all co-authors as well as by the responsible authorities at the institute where the work has been carried out. The publisher will not be held legally responsible should there be any claims for compensation.
Competing interests
The authors declare that they have no competing interests.
References
-
Clemen
C.S.,
Herrmann
H.,
Strelkov
S.V.,
Schröder
R.,
Desminopathies: pathology and mechanisms. Acta Neuropathologica.
2013;
125
(1)
:
47-75
.
View Article PubMed Google Scholar -
Ripoll-Vera
T.,
Zorio
E.,
Gámez
J.M.,
Molina
P.,
Govea
N.,
Crémer
D.,
Phenotypic Patterns of Cardiomyopathy Caused by Mutations in the Desmin Gene. A Clinical and Genetic Study in Two Inherited Heart Disease Units. Rev Esp Cardiol (Engl Ed).
2015;
68
(11)
:
1027-9
.
View Article PubMed Google Scholar -
Schröder
R.,
Schoser
B.,
Myofibrillar myopathies: a clinical and myopathological guide. Brain Pathology (Zurich, Switzerland).
2009;
19
(3)
:
483-92
.
View Article PubMed Google Scholar -
van Spaendonck-Zwarts
K.Y.,
van Hessem
L.,
Jongbloed
J.D.,
de Walle
H.E.,
Capetanaki
Y.,
van der Kooi
A.J.,
Desmin-related myopathy. Clinical Genetics.
2011;
80
(4)
:
354-66
.
View Article PubMed Google Scholar -
Olivé
M.,
Goldfarb
L.,
Moreno
D.,
Laforet
E.,
Dagvadorj
A.,
Sambuughin
N.,
Desmin-related myopathy: clinical, electrophysiological, radiological, neuropathological and genetic studies. Journal of the Neurological Sciences.
2004;
219
(1-2)
:
125-37
.
View Article PubMed Google Scholar -
Brodehl
A.,
Gaertner-Rommel
A.,
Milting
H.,
Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophysical Reviews.
2018;
10
(4)
:
983-1006
.
View Article PubMed Google Scholar -
Azzimato
V.,
Gennebäck
N.,
Tabish
A.M.,
Buyandelger
B.,
Knöll
R.,
Desmin, desminopathy and the complexity of genetics. Journal of Molecular and Cellular Cardiology.
2016;
92
:
93-5
.
View Article PubMed Google Scholar -
Pawlak
A.,
Rejmak-Kozicka
E.,
Gil
K.E.,
Ziemba
A.,
Kaczmarek
L.,
Gil
R.J.,
Patterns of desmin expression in idiopathic dilated cardiomyopathy are related to the desmin mRNA and ubiquitin expression. Journal of Investigative Medicine.
2019;
67
(1)
:
11-9
.
View Article PubMed Google Scholar -
Harada
H.,
Hayashi
T.,
Nishi
H.,
Kusaba
K.,
Koga
Y.,
Koga
Y.,
Phenotypic expression of a novel desmin gene mutation: hypertrophic cardiomyopathy followed by systemic myopathy. Journal of Human Genetics.
2018;
63
(2)
:
249-54
.
View Article PubMed Google Scholar -
Brodehl
A.,
Pour Hakimi
S.A.,
Stanasiuk
C.,
Ratnavadivel
S.,
Hendig
D.,
Gaertner
A.,
Restrictive cardiomyopathy is caused by a novel homozygous desmin (DES) mutation p.Y122H leading to a severe filament assembly defect. Genes.
2019;
10
(11)
:
918
.
View Article PubMed Google Scholar -
Kubánek
M.,
Schimerová
T.,
Piherová
L.,
Brodehl
A.,
Krebsová
A.,
Ratnavadivel
S.,
Desminopathy: novel desmin variants, a new cardiac phenotype, and furder evidence for secondary mitochondria dysfunction. Journal of Clinical Medicine.
2020;
9
(4)
:
937
.
View Article PubMed Google Scholar -
Tamiya
R.,
Saito
Y.,
Fukamachi
D.,
Nagashima
K.,
Aizawa
Y.,
Ohkubo
K.,
Desmin-related myopathy characterized by non-compaction cardiomyopathy, cardiac conduction defect, and coronary artery dissection. ESC Heart Failure.
2020;
7
(3)
:
1338-43
.
View Article PubMed Google Scholar -
McLaughlin
H.M.,
Kelly
M.A.,
Hawley
P.P.,
Darras
B.T.,
Funke
B.,
Picker
J.,
Compound heterozygosity of predicted loss-of-function DES variants in a family with recessive desminopathy. BMC Medical Genetics.
2013;
14
(1)
:
68
.
View Article PubMed Google Scholar -
Henderson
M.,
De Waele
L.,
Hudson
J.,
Eagle
M.,
Sewry
C.,
Marsh
J.,
Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathologica.
2013;
125
(6)
:
917-9
.
View Article PubMed Google Scholar -
Durmuş
H.,
Ayhan
Ö.,
Ç\irak
S.,
Deymeer
F.,
Parman
Y.,
Franke
A.,
Neuromuscular endplate pathology in recessive desminopathies: lessons from man and mice. Neurology.
2016;
87
(8)
:
799-805
.
View Article PubMed Google Scholar -
Linthorst
G.E.,
Hollak
C.E.,
[Whole exome sequencing and whole genome sequencing in undiagnosed disease: of value for certain patient populations]. Nederlands Tijdschrift voor Geneeskunde.
2019;
163
:
3711
.
PubMed Google Scholar -
Sun
Y.,
Man
J.,
Wan
Y.,
Pan
G.,
Du
L.,
Li
L.,
Targeted next-generation sequencing as a comprehensive test for Mendelian diseases: a cohort diagnostic study. Scientific Reports.
2018;
8
(1)
:
11646
.
View Article PubMed Google Scholar -
Barbitoff
Y.A.,
Polev
D.E.,
Glotov
A.S.,
Serebryakova
E.A.,
Shcherbakova
I.V.,
Kiselev
A.M.,
Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Scientific Reports.
2020;
10
(1)
:
2057
.
View Article PubMed Google Scholar -
Choi
M.,
Scholl
U.I.,
Ji
W.,
Liu
T.,
Tikhonova
I.R.,
Zumbo
P.,
Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America.
2009;
106
(45)
:
19096-101
.
View Article PubMed Google Scholar -
Ng
S.B.,
Turner
E.H.,
Robertson
P.D.,
Flygare
S.D.,
Bigham
A.W.,
Lee
C.,
Targeted capture and massively parallel sequencing of 12 human exomes. Nature.
2009;
461
(7261)
:
272-6
.
View Article PubMed Google Scholar -
Bär
H.,
Strelkov
S.V.,
Sjöberg
G.,
Aebi
U.,
Herrmann
H.,
The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation?. Journal of Structural Biology.
2004;
148
(2)
:
137-52
.
View Article PubMed Google Scholar -
Singh
S.R.,
Kadioglu
H.,
Patel
K.,
Carrier
L.,
Agnetti
G.,
Is desmin propensity to aggregate part of its protective function?. Cells.
2020;
9
(2)
:
491
.
View Article PubMed Google Scholar -
Smolina
N.,
Khudiakov
A.,
Knyazeva
A.,
Zlotina
A.,
Sukhareva
K.,
Kondratov
K.,
Desmin mutations result in mitochondrial dysfunction regardless of their aggregation properties. Biochimica et Biophysica Acta. Molecular Basis of Disease.
2020;
1866
(6)
:
165745
.
View Article PubMed Google Scholar -
Schröder
R.,
Goudeau
B.,
Simon
M.C.,
Fischer
D.,
Eggermann
T.,
Clemen
C.S.,
On noxious desmin: functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria. Human Molecular Genetics.
2003;
12
(6)
:
657-69
.
View Article PubMed Google Scholar -
Wahbi
K.,
Béhin
A.,
Charron
P.,
Dunand
M.,
Richard
P.,
Meune
C.,
High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: a 10-year longitudinal study. Neuromuscular Disorders.
2012;
22
(3)
:
211-8
.
View Article PubMed Google Scholar -
Vrabie
A.,
Goldfarb
L.G.,
Shatunov
A.,
Nägele
A.,
Fritz
P.,
Kaczmarek
I.,
The enlarging spectrum of desminopathies: new morphological findings, eastward geographic spread, novel exon 3 desmin mutation. Acta Neuropathologica.
2005;
109
(4)
:
411-7
.
View Article PubMed Google Scholar -
Goldfarb
L.G.,
Vicart
P.,
Goebel
H.H.,
Dalakas
M.C.,
Desmin myopathy. Brain.
2004;
127
(Pt 4)
:
723-34
.
View Article PubMed Google Scholar -
Goldfarb
L.G.,
Park
K.Y.,
Cervenáková
L.,
Gorokhova
S.,
Lee
H.S.,
Vasconcelos
O.,
Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nature Genetics.
1998;
19
(4)
:
402-3
.
View Article PubMed Google Scholar -
Brodehl
A.,
Dieding
M.,
Klauke
B.,
Dec
E.,
Madaan
S.,
Huang
T.,
The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circulation: Cardiovascular Genetics.
2013;
6
(6)
:
615-23
.
View Article PubMed Google Scholar -
Park
K.Y.,
Dalakas
M.C.,
Goebel
H.H.,
Ferrans
V.J.,
Semino-Mora
C.,
Litvak
S.,
Desmin splice variants causing cardiac and skeletal myopathy. Journal of Medical Genetics.
2000;
37
(11)
:
851-7
.
View Article PubMed Google Scholar
Comments
Downloads
Article Details
Volume & Issue : Vol 8 No 12 (2021)
Page No.: 4809-4817
Published on: 2021-12-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 6395 times
- PDF downloaded - 991 times
- XML downloaded - 0 times
- Supplement downloaded - 1097 times
