

Copyrights: Garima Sharma, Badruddeen ., Juber Akhtar, Mohammad Irfan Khan, Mohammad Ahmad, Deepa Neopane, 2024. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Diabetic neuropathy (DN) is a debilitating complication of diabetes mellitus (DM), characterized by nerve damage resulting from chronic hyperglycemia. This condition affects a significant proportion of diabetic patients, leading to symptoms such as weakness, numbness, and pain, particularly in the extremities. The pathogenesis of DN is complex and involves metabolic, vascular, and neurotrophic factors. At the core of its development are various receptors that mediate and modulate the underlying biochemical and cellular processes. Key receptors implicated in DN include the advanced glycation end-product receptor (RAGE), which is involved in oxidative stress and inflammation. Additionally, transient receptor potential channels, namely TRP channels, particularly TRPV1 and TRPA1, play an important role in the sensation of pain and thermal stimuli, contributing to the sensory abnormalities observed in DN. Insulin and insulin-like growth factor receptors also play significant roles, as insulin signaling is crucial for neuronal survival and function. Furthermore, purinergic receptors, specifically the P2X and P2Y subtypes, are involved in neuroinflammation and pain transmission. Understanding the roles and interactions of these receptors provides valuable insights into the pathophysiology of DN and highlights potential therapeutic targets. Future research focusing on modulating these receptor pathways holds promise for developing effective treatments to alleviate symptoms and potentially reverse the progression of DN.
Introduction
DN is one of the most chronic and prevalent conditions described as supplementary nerve dysfunction symptoms in cases with DM, affecting over 50% of people with diabetes. According to data recently issued by the International Diabetes Federation (IDF), an estimated 537 million individuals worldwide currently have diabetes. By 2030 and 2045, that number is projected to rise to 643 million and 783 million, respectively, a significant cause of DN1, 2, 3. DN causes a variety of clinical symptoms, including pain and sensory loss. It also increases the chance of foot ulcers and amputation, an irreversible consequence for cases. Positive symptoms, such as painful DN, are endured by one-third of people with neuropathy and include paraesthesia, allodynia, and spontaneous pain. DN might appear as sensory, motor, focal/multifocal, or autonomic neuropathies. Diabetic distal symmetric polyneuropathy (DSPN) is the most common type of DN, making up over 75% of all diabetic neuropathies. The distal terminals of sensory neurons are gradually and stealthily damaged in DSPN cases, resulting in symptoms similar to tingling, discomfort, or lack of sensation in their toes. Because nerve damage and discomfort take longer to display in older persons (over 50), diabetic neuropathy is more common in this population. As per the Search for Diabetes in Youth study, the prevalence of peripheral DN in children up to the age group of 20 years is approximately 7% (those with type-I diabetes) and up to 22% (with type-II diabetes) respectively4, 5, 6, 7. Molecular neurobiological knowledge of human nervous tissues is required to produce the next generation of therapeutics for neurological disorders like chronic pain. Significant gene families and pathways were analyzed, including transcription factors (TFs), G-protein-coupled receptors (GPCRs), & ion channels8. Peripheral sensory neurons in the dorsal root ganglion (DRG) & trigeminal ganglion (TG) are specialized to detect & transduce different natural stimuli including touch, temperature, pain, etc., to the CNS. The recent resources and information available can guide future studies in comparative transcriptomics, simplify cell-type terminological differences across studies, and offer assistance in prioritizing targets for future pain treatment advancements9. Sensory neurons of the DRG are essential for maintaining tissue homeostasis by detecting & initiating reactions to stimuli. Whereas most preclinical studies of DRGs are conducted on rodents, much less is known about the mechanisms of sensory recognition in primates10. Pregabalin and duloxetine showed significant therapeutic impacts on painful DPN, but adverse events were too significant. The pain-relieving effects of ABT-894 and gabapentin need to be further studied with longer and larger RCTs. As an opioid drug, tapentadol has a significant pain-relieving impact, but due to its addiction potential, it needs to be used cautiously in clinical practice. Pregabalin, duloxetine, & tapentadol, three drugs are approved by the US Food and Drug Administration (USFDA) for the DPN treatment. Although lacosamide, mirogabalin, and capsaicin are more effective than placebo, their therapeutic impact is weaker than that of pregabalin. For satisfactory results, long-term studies are still required to confirm their efficacy and safety in the future11.
MECHANISM AND PATHOPHYSIOLOGY
As noted, DN is a distinct neurodegenerative condition affecting the peripheral nervous system, firstly impacting autonomic axons, sensory axons, and lower limb motor axons. It's unclear how DM affects sensory neurons. In progressive DN, the perikarya (cell bodies) are largely preserved while the terminal sensory axons in the periphery shrink and die back. DN is classified as a length-dependent neuropathy due to its involvement pattern, which resembles a "stocking & glove" pattern. According to this pattern, the longest sensory axons are first affected, with loss of distal leg epidermal axons occurring before loss in more proximal limbs. Schwann cells are targets in chronic hyperglycemia. Moreover, severe cases of DN in individuals involve features of demyelination, although DN is not considered to be predominantly a demyelinating neuropathy, since axons and Schwann cells may induce various changes in the axon. For instance, Schwann cells are crucial in controlling the cytoskeletal characteristics of axons, such as the parameters governing the trafficking of axons and the location of proteins at Ranvier's nodes. Faulty Schwann cell-axon ribosome transfer, inadequate cytoskeletal support, or lack of trophic factors, facilitate the intra-axonal translation of mRNA within the distal axons. Ribosome-filled Schwann cells in mice can regulate the synthesis of axonal proteins when introduced to demyelinated axons. When axons are under pressure or damaged, this ribosome transfer may make axon-Schwann cell connections even more pivotal12, 13.
It's uncertain whether diabetes triggers innate axonal programs that contribute to axonal deterioration. Exploration of Wallerian degeneration has revealed intracellular signaling pathways that activate axonal degeneration. One of these pathways’ major regulators appears to be mononucleotide adenylyl transferase (NMNAT), often referred to as NMN/NaMN adenylyl transferase. However, it's still unclear if diabetes activates these pathways14. Axon changes, particularly those affecting the distal terminals, are associated with changes in the neuronal perikarya. DRG sensory neurons change their phenotype in long-term experimental diabetes, which may be crucial for how they maintain the branches of distant axons. For example, the production and export of neurofilament polymers, essential for maintaining the structural scaffolding of the axon, are gradually lost in rats with chronic Type-1 Diabetes. This loss of neurofilament polymers has been suggested to be caused by reduced mRNA expression encoding neurofilament. Endoplasmic reticulum stress is also implicated in preclinical research with diabetic animals in diabetes-induced peripheral nerve injury that could affect nerve functions. Similarly, research conducted in vivo and in vitro in rodent models has shown that the expression patterns of heat shock proteins (HSPs) and poly (ADP-ribose) polymerase (PARP) in DRGs, as well as the function of synaptic plasticity molecules like growth-associated protein (GAP43 or known as neuromodulin) and β-tubulin, are altered due to hyperglycemia. Data indicate that in these pathways, any dysfunction promotes the processing of abnormal protein, oxidative damage, and mitochondrial dysfunction, leading to loss of peripheral nerve function, even when the mechanisms of damage are still being investigated.
This notion is supported by the observation that nerve function can be improved by modifying individual molecules in these pathways. For example, nerve conduction velocity (NCV) and responses to mechanical and thermal stimuli, both clinically relevant endpoints, were improved by controlling HSP90 activation, most likely as a result of enhanced mitochondrial function. Recent array studies have similarly demonstrated that DRG sensory neurons exposed to chronic diabetes exhibit a variety of mRNA and microRNA alterations. Preclinical models of Type-I DM and Type-II DM have shown increased expression of inflammation, bioenergetic, and lipid processing-related pathways in a variety of sciatic nerve cultures (Figure 1)15, 16, 17, 18, 19, 20.
Furthermore, a study comparing gene expression patterns in peripheral nerves from individuals with Type-I DM and Type-II DM with those from mouse models of DN identified several largely conserved pathways related to inflammation, lipid metabolism, and adipogenesis21. DN has also been associated with other specific changes in the DRG and nerve function, such as altered spliceosome function, changes in the production of survival motor neuron protein, and overexpression of GW-bodies (mRNA processing sites)22.

| No | Receptor | Sub-types | Class | Distribution body | References |
|---|---|---|---|---|---|
| 1 | Transient receptor potential channels (TRP channels) | *TRPC(Canonical): TRPC1, TRPC2, TRPC3, TRPC6, TRPC7, TRPC4, TRPC5. *TRPV (Vanilloid), | Six Trans-Membrane Helical Voltage-Gated Ion Channels, cation permeable plasma membrane channels with varying Ca 2+ selectivity | Embryonic brain, liver, kidney tissues as well as adult heart, testes, ovaries, brain, Sensory Neuron, Plasma membrane, Intracellular Compartment | 23 , 24 25 , 26 27 28 |
| 2 | Toll-Like Receptors | *V type-TLR | Membrane-Bound Receptor | Macrophages, dendritic cells (DCs), natural killer cells (NKs), mast cells, basophils, eosinophils (specialized immune cells) as well as normal cells etc . | 29 , 30 , 31 , 32 |
| 3 | Insulin Receptors | IR-A IR-B | Transmembrane Protein & Part of Tyrosine Kinase Receptor (Rtk) | Pancreatic beta cells, some areas of CNS, Adipocytes, Myocytes, Hepatocytes, Skeletal Muscle | 33 , 34 , 35 |
| 4 | N-methyl-D- aspartate receptor | GluN1/GluN2A GluN1/GluN2B GluN1/GluN2C GluN1/GluN2C | Ligand-Gated Ion Channels | Hippocampus, Spinal Cord, Neocortex, Cerebellum | 36 , 37 , 38 |
| 5 | Opioid Receptors | m (mu): MOP, d (delta): DOP, k (kappa): KOP, FQ (N/OFQ) or NOP | G protein-coupled receptors (GPCR) | In CNS: Locus coeruleus, medulla, periaqueductal grey area, Midbrain, Limbic- Cortical Structures, Cerebellum, Caudate Nucleus, Nucleus Accumbens, Hippocampus, Cerebral Cortex, Putamen, Temporal Lobe, Hypothalamic Nuclei. In the periphery: neuronal & non-neuronal tissues including (neuroendocrine), immune cells, ectodermal cells, and smooth muscle cells & at the terminal of sympathetic & sensory peripheral neurons | 39 , 40 , 41 , 42 , 43 |
| 6 | AGE-RAGE receptor | *Full-length RAGE (fl-RAGE) *N-Truncated RAGE *Dominant -ve RAGE *Soluble RAGE (sRAGE) *Soluble RAGE derived from proteolytic cleavage | Transmembrane Multi-Ligand Receptor of Immunoglobulin Superfamily | Epithelial vascular endothelial cells, immune cells, monocytes, macrophages, neurons, cardiomyocytes, adipocytes, glomerular epithelial cells, podocytes, and alveolar epithelial cells | 44 , 45 , 46 , 47 |
| 7 | Purinoceptors (P2Y, P2X) | *P1: A1, A2A, A2B, A3 *P2: P2X seven subunits (P2X1 to P2X7), P2Y eight subtypes (P2Y1, P2Y2, P2Y4, P2Y6, & P2Y11 to P2Y14 | G-protein coupled receptor (GPCR) (P1), Ligand-Gated Ion Channel- (P2X) | P1: brain, autonomic nerve terminal, spinal cord, heart, spleen, lungs P2: Central Nervous System, Smooth Muscle, Dorsal Horn, Pancreas, Spinal Neurons, Endothelial Cells, Chromaffin Cells etc . | 48 , 49 |
| 8 | Nerve Growth Factor (NGF) Receptors | Trk: TrkA, TrkB, TrkC p75: p75NTR | Tumor Necrosis Factor (TNF) Trans Membrane Receptor Superfamily | Hippocampus, cortex, basal ganglia, thalamus, spinal cord, retina | 50 , 51 , 52 |
| 9 | Glutamate Receptors | AMPK, KAINATE, NMDA, GluD | Cation-permeable ligand-gated ion channels (ionotropic glutamate receptor), G protein-coupled (metabotropic receptor) | Membranes Of Neuronal And Glial Cells, Human Brain, Fusiform Cells Of The Dorsal Cochlear Nucleus (Cn), Basal Dendrite Synapses (Auditory Nerve) | 53 , 54 , 55 |
| 10 | Cannabinoid receptors | CB1 & CB2 | G protein-coupled receptor Super- Family | Central Nervous System, Brain, Microglia, Immune Cells | 56 , 57 , 58 |
| 11 | Serotonin Receptors (5HT) | 5HT1, 5HT2, 5HT3, 5HT4, 5HT6, 5HT7 | Ligand gated ion channel and G protein coupled receptor | Gastro Intestinal tract, Central Nervous System, Platelets, Cardiovascular System, Immune Cells | 59 , 60 |
| 12 | Adrenergic Receptors | α: α1 (α1A, α1B, α1D), α2 (α2A, α2B, α2C), | G protein coupled Receptor | Central Nervous System, Peripheral Nervous System, Blood Vessels, Platelets, Nerve Terminals, Heart, Smooth Muscles, Lungs etc . | 61 , 62 , 63 , 64 |
| 13 | Bradykinin (BK) Receptors | B1R B2R | G Protein Coupled Receptor | Cardiovascular System, Brain, Kidney, Nerve Fibers, Lamina Propria of Tissue Linin of Pelvis etc . | 65 , 66 , 67 |
| 14 | Chemokine Receptor | CC, CXC, CX3C, XC | G Protein Coupled Receptor | Neutrophils, Leukocytes & Immune Cells, Dendritic Cells, Endothelial Cell | 68 , 69 , 70 |
| 15 | Muscarinic receptor | M1, M2, M3, M4, M5 | G protein coupled Receptor | Brain, Prostate, Salivary Gland, Heart, Bladder, Intestine, Lung, Pancreas etc . | 71 , 72 , 73 |
RECEPTORS AND THEIR ROLES IN DIABETIC NEUROPATHY
Diabetic peripheral neuropathy (DPN) involves several classes of receptors that contribute to its pathophysiology. Various mechanisms like pain, sensation, inflammation, neural functions, etc., are mediated through these receptors. Table 1 lists receptors involved in diabetic neuropathy, detailing their class, subtype, and location in the body.
TRANSIENT RECEPTOR POTENTIAL (TRP) CHANNELS
Ten people experience neuropathic pain on a chronic basis, representing a significant public health burden that affects social resources and quality of life. Numerous pathophysiological conditions, such as cancer treatment, spinal cord injuries, viral infections, and multiple sclerosis, in addition to diabetes, can lead to persistent neuropathic pain. Specialized afferent sensory neurons known as nociceptors are activated when chemical, mechanical, and heat stimuli exceed a certain threshold. These neurons innervate peripheral organs & tissues with their cell bodies located in the trigeminal ganglia and dorsal root, projecting centrally into the brain stem and spinal cord, respectively. The primary and earliest symptoms of neuropathic pain are hyperalgesia (increased pain from a stimulus that normally provokes pain) and allodynia (pain due to a stimulus that does not typically evoke pain). These symptoms are characterized by sensitization of the pain circuits from the nociceptors to the brain. The Transient Receptor Potential family of ion channels, expressed on nociceptors, was identified as a significant discovery in the search for mechanisms of nociception. Their crucial roles in nociception offer new targets for treating clinical neuropathic pain conditions. TRP genes have been identified since the nociceptive TRPV1, the capsaicin receptor, was characterized in the late 1990s. Their products are classified into 6 subfamilies: TRPV (Vanilloid), TRPC (Canonical), TRPM (Melastatin), TRPA (Ankyrin), TRPP (Polycystin), and TRPML (Mucolipin). Individuals with diabetes exhibit biphasic pain behaviors, with hyperalgesia during the initial phase of the disease and transitioning to hypoalgesia in later stages. During the hyperalgesia phase, TRPV1 expression was found to be upregulated in sensory inputs from the spinal cord, DRG somata, and skin-innervating axons. Increased TRPV1 currents seen from isolated DRG neurons were associated with this elevated expression. The absence of diabetes-induced heat hypersensitivity in TRPV1-deficient animals and alleviation of diabetes-induced mechanical allodynia in mice, administered a TRPV1 antagonist intrathecally, validated TRPV1's role in the hyperalgesic phase of the disease. Hyperglycemia and hypoxia-induced sensitization of TRPV1 channels, recurrently associated with the early stages of diabetic pain, might be a presumptive explanation for this hypersensitivity. On the other hand, in both humans and animals, the disease's late stage is marked by a loss of sensitivity. Studies involving diabetic patient samples have revealed a decrease in TRPV1 expression in skin-embedded nerve terminals. Similar findings have been observed in animal models during the advanced stages of the disease when hypoalgesic mice exhibited reduced expression of TRPV1. Additionally, it has also been shown through calcium imaging and electrical recordings that DRG neurons derived from diabetic hypoalgesic rats exhibit reduced channel activity. Mechanistically, hypoalgesia could be mediated by resistance to or even loss of insulin signaling in DRGs, occurring in the late phase of diabetes. This hypothesis is supported by evidence showing that insulin enhances TRPV1 activity in DRG neurons74, 75, 76, 77, 78.
Chemotherapy and diabetes-related peripheral neuropathies have been shown to be influenced by TRPA1. Inhibiting TRPA1 channels in diabetic animals reduces the mechanical hypersensitivity observed in the earliest phase of the disease. In the advanced stages of the illness, the activation of TRPA1 mediates the loss of substance P-containing filaments, resulting in changes in tactile sensitivity. The functionality of these filaments may be restored by pharmacologically blocking the channel. "Methylglyoxal" (MG), an intracellular derivative of several metabolic processes such as lipid peroxidation and glycolysis, becomes elevated in concentration in the plasma and intracellular milieu of patients with diabetes due to hyperglycemia. Recent research indicates that MG directly activates TRPA1 by binding to the channel's intracellular region. Consequently, MG injections in the hind paw induce pain in a manner dependent on TRPA1, and pharmacological blockade of the channel reduces diabetic neuropathic pain. Additionally, both diabetes and chemotherapy-induced neuropathic pain have been linked to the function of the osmosensor TRPV4 channel. Rats given intrathecal TRPV4 siRNA or TRPV4 null mice exhibited a reduction in mechanical hypersensitivity under these conditions74, 75, 76, 77, 78, 79, 80, 81, 82.
TOLL-LIKE RECEPTORS
Toll-like receptors, a class of proteins, are crucial to the innate immune response. Interactions between TLR ligands and microbial pathogen-expressed ligands can initiate a signaling cascade that produces cytokines and triggers an adaptive immune response. TLR4 is one of the TLRs involved in numerous inflammatory complications. Research involving both human subjects and animal models suggests that TLR4-mediated systemic inflammation plays a role in the pathophysiological process of diabetes. Likewise, data from mice indicate that activation of TLR2 and TLR4, along with subsequent cytokine synthesis, may contribute to diabetes progression. Studies of human and animal models have also revealed a connection between diabetes and alterations in the innate immune system. TLR4, an essential innate immune receptor, can identify pathogens, initiate an inflammatory response, and trigger an adaptive immunological reaction. Increased TLR4 mRNA expression has been observed in the secreting adipose tissue of db/db mice, and there is a significant correlation between elevated TLR4 expression and TLR-mediated inflammation in monocytes and HbA1c levels in diabetes patients. Research has demonstrated that TLR4 deficiency in mice reduces the proinflammatory state associated with diabetes, and TLR4 genotypes Asp299Gly and/or Thr399Ile are related to a lower prevalence of diabetic nephropathy in individuals with type 2 diabetes. Collectively, these findings suggest a potential involvement of TLR4 in the pathophysiology of diabetes and DPN. Two downstream pathways that TLR4 can activate within a cell are the mitogen-activated protein kinase (MAPK) and NF-κB pathways. While activation of the MAPK pathway is crucial for controlling neural plasticity, activation of the NF-κB pathway results in the production and release of proinflammatory cytokines, such as TNFα and IL-6. Experiments with inhibitors of the IKK (inhibitory κB kinase) pathways and peripheral blood mononuclear cells have demonstrated the critical role of NF-κB in regulating insulin response, glucose metabolism, and inflammatory responses. TNF-α and IL-6 are key players in endothelial cell permeability, proliferation, and matrix overproduction. They also affect the activity of glial cells and neurons, which is related to the pathogenic processes of DN and diabetic retinopathy (DR). It has been found that TNF-α inactivation helps improve DN in mice83, 84, 85, 86, 87, 88.
INSULIN RECEPTORS
Type-I and type-II diabetes mellitus (DM) lead to impairments in insulin signaling in the peripheral nervous system, contributing to the DPN phenotype. The interaction between insulin and the insulin receptor (IR) is necessary for insulin signaling. This interaction causes the IR to auto-phosphorylate on intracellular tyrosine residues, activating intracellular signaling pathways. The IR is expressed in neurons and glia of both the central and peripheral nervous systems. Notably, IRs are concentrated in the nodal areas of Schwann cells and axonal membranes. It has been shown that treatment with insulin inhibits hyperglycemia-induced mitochondrial depolarization in neurons and that insulin localizes to mitochondria in neurons. Additionally, several DPN phenotypes in animals were partially reversed by intrathecal insulin administration or near-sciatic nerve electrical stimulation. Diabetes affects both neurotrophic support and neural regeneration of damaged axons, two processes in which insulin and other growth factors play essential roles. Moreover, recent research has demonstrated a reduced ability of cultured dorsal root ganglion (DRG) neurons to respond to acute insulin stimulation when previously exposed to chronic insulin stimulation, a model of type-II DM hyperinsulinemia. The diminished sensitivity of DRG neurons to insulin-induced Akt activation is one of the mechanisms underlying part of this insulin resistance. These findings indicate that neurons, similarly to traditional "insulin-sensitive" tissues, can develop insulin resistance. In cases of neuropathy or neuronal injury, neurons may have limited regeneration capability due to their reduced responsiveness to the neurotrophic effects of insulin under hyperinsulinemia conditions. Given their previously mentioned role in axonal support, it would be pertinent to investigate whether Schwann cells experience insulin resistance under conditions of diabetes89, 90, 91, 92.
N-METHYL-D-ASPARTATE RECEPTORS/NMDA RECEPTORS
Two major complaints from individuals with DN are allodynia and hyperalgesia. In DN, the spinal cord's dorsal horn plays a significant role in nociceptive signal perception. Increased primary afferent activity may contribute to central sensitization or hyperactivity of dorsal horn neurons in the spinal cord, as well as neuroplasticity. NMDA receptors are heteromeric protein complexes. It has been demonstrated that activation of spinal NMDA receptor activity leads to hyperalgesia in neuropathic pain conditions. For example, compared to control rats, nerve-ligated rats exhibit a significant increase in Ca2+ influx in spinal lamina II neurons upon administration of NMDA. In rats with spinal nerve ligation, the NMDA receptor antagonists ketamine, memantine, or MK 801 consistently decrease elicited responses from dorsal horn neurons. Additionally, in animals with nerve ligation, the amplitude of NMDA receptor currents is reduced by the NR2B subunit-specific antagonist ifenprodil. However, accelerated glutamate release from the primary afferent terminals of the spinal cord is associated with DN. The GABA-B receptor is highly expressed at the dorsal root of the spinal cord, and activation of this receptor produces antinociception effects in both acute and chronic pain. Studies have shown that in the spinal dorsal horn, presynaptic GABA-B receptor stimulation reduces synaptic release of GABA, glycine, and glutamate, and postsynaptic GABA-B receptor activation decreases the Ca2+ permeability of NMDA receptors to reduce Ca2+ signals in postsynaptic spines, in addition to suppressing the release of presynaptic neurotransmitters. GABA-B receptors are distributed pre- and postsynaptically in the dorsal horn of the spinal cord. They are G-protein-coupled receptors. Through their inhibition of Ca2+ channels or interactions with downstream release machinery, presynaptic GABA-B receptors deplete neurotransmitter release. Through direct inhibition of Ca2+ channels (voltage-sensitive) and the opening of inwardly rectifying K+ channels, postsynaptic GABA-B receptors reduce neuronal excitability. Slow inhibitory postsynaptic potentials are produced when these K+ channels are opened, strengthening the magnesium blockade of NMDA receptors. The mechanism behind the GABA-B receptor’s inhibitory influence on Ca2+ signals in dendrites and spines is the indirect block of NMDA receptors. GABA-B activation leads to downregulation of NMDA receptor expression in the spinal cord at both mRNA and protein levels. Various noncompetitive NMDA receptor antagonists (i.e., MK-801, ketamine, memantine, and dextrorphan) decrease the development of allodynia and hyperalgesia following constrictive injury of the sciatic nerve and spinal nerve ligation. Moreover, the intrathecal application of amino-5-phosphonopentanoate (a competitive NMDA receptor antagonist) reduces mechanical allodynia caused by spinal cord damage. Parenteral and oral administration of norketamine (an important metabolite of ketamine) alleviates mechanical and thermal hyperalgesia, but not tactile allodynia, in the sciatic nerve injury model without significant side effects. Compared with ketamine and MK-801, the low-affinity NMDA receptor blockers memantine and neramexane exhibit faster unblocking kinetics and more pronounced voltage dependency. Thus, these agents may be able to hinder the ongoing NMDA receptor activation presumed to occur in neuropathic pain with a lower incidence of side effects. Chronic administration of neramexane and memantine (noncompetitive NMDA receptor antagonists) for 2 weeks produces persistent antinociceptive effects on mechanical hyperalgesia and allodynia in a rodent model of DN pain. Several studies have illustrated that specific glycine-site NMDA receptor antagonists, such as L-701,324, MRZ2/576, and 5,7-dichlorokinurenic acid (5,7-DCK), reverse allodynia induced. GV196771 and Pyridazinoquinolinetriones, modern glycine-site NMDA receptor antagonists (with improved oral bioavailability), can decrease neuropathic pain induced by sciatic nerve damage in rats93, 94.
OPIOID RECEPTORS
Opioid receptors, which are G-protein coupled receptors, are categorized into mu (MOR), delta (DOR), and kappa (KOR). These receptors block the transmission of pain at various stages of the ascending pain pathways and in supraspinal areas associated with pain integration. Additionally, opioid receptors are involved in descending pathways that exert both facilitative and inhibitory effects, initially affecting the rostral ventromedial medulla and the periaqueductal grey. The majority of the pharmacological effects of currently available opioids are produced through the activation of MOR, and the opioids used to manage pain are referred to as "mu agonists." The importance of opioid receptor complexes in pain attenuation in diabetic neuropathy is also well recognized. The spinal cord plays a crucial role in the transduction of pain. Mu opioid receptors (MORs) are found in the presynaptic central terminals of primary afferent neurons, a key site for pain signal transmission. These neurons are the targets of drugs prescribed for neuropathic pain, similar to gabapentinoids, as well as opioids administered via spinal injection. Such analgesics block transmitters that facilitate the brain's processing of pain by inhibiting voltage-gated calcium channels. Since opioid receptors are located on the postsynaptic membrane of primary afferent neurons and the presynaptic membrane of secondary afferent neurons, their activation disrupts the release of neurotransmitters and the transmission of peripheral signals to the brain. Furthermore, there was a study that investigated the antiallodynic, antinociceptive, and anti-hyperalgesic effects of morphine, fentanyl, and a new opioid agonist, "14-O-methylmorphine-6-O-sulfate" (14-O-MeM6SU), in rats with streptozotocin-induced diabetic neuropathy pain (DNP) after systemic treatment. When comparing diabetic rats to non-diabetic rats, only "14-O-MeM6SU" showed antiallodynic properties at dosages equivalent to the antinociceptive levels observed in non-diabetic rats. In diabetic rats, there was a decrease in the number of spinal mu opioid receptor binding sites and a reduction in MOR immunoreactivity in nerve terminals in the spinal cord as well as in the dorsal root ganglia. At the spinal/supraspinal levels, the G-protein coupling assay revealed a decreased efficacy profile for 'morphine' and a high efficacy profile for 'fentanyl' or '14-O-MeM6SU'. Also, in tissues from both diabetic and non-diabetic rats, only '14-O-MeM6SU' demonstrated equivalent efficacy in G-protein activation at the spinal level. This suggests that the loss of MOR, particularly in the spinal cord, might be the reason for the reduced analgesic effect of opioids in diabetic neuropathy. Therefore, there is a need to develop broadly effective opioids that may offer analgesia superior to that of opioids currently available on the market for effectively treating diabetic peripheral neuropathy95, 96.
AGE-RAGE RECEPTOR
In a hyperglycemic environment, tissue glucose levels rise, which in turn causes the peripheral nervous system to produce AGEs (Advanced Glycation End-products). Diabetes greatly facilitates the production and accumulation of AGEs because glucose serves as the primary source of carbonyl groups for glycation processes. Additionally, pyrraline, N-carboxymethyl lysine, methylglyoxal/glyoxal lysine, and pentosidine are the main AGEs in vivo. Methylglyoxal, a substance generated from glucose, modifies proteins, aiding in the development of AGEs. This common substance in diabetic tissues is broken up by the enzyme glyoxalase 1 (a rate-limiting enzyme), which is abundantly expressed in hyperglycemic conditions. The localization of AGEs has been researched in both human and experimental diabetic animal peripheral nerves. AGE receptors may play a part in the emergence of diabetes complications, in addition to the buildup of AGEs in tissues. AGEs have been shown to bind to several receptors, such as the AGE receptor (RAGE), scavenger receptor class A (SR-A), galectin-3, lox-1, CD36, and SR-BI; the latter three are components of scavenger receptor class B.
The peripheral nerve is altered both structurally and functionally when proteins are modified by AGEs. It has been demonstrated that when AGEs were added to neuronal cells and Schwann cells, the cells died in vitro. The accumulation of AGEs may contribute to the loss of fiber in the diabetic peripheral nerve in humans. The AGE-induced alteration of neurofilament and tubulin may also obstruct axonal transport. The progression of atrophy and deterioration of nerve filaments is subsequently facilitated by the disruption of axonal transport. The surface protein known as a receptor for AGE (RAGE) has a mass of 45–55 kDa. Its structure allows us to pinpoint three distinct fractions, each serving a specific purpose. RAGE is a multi-ligand transmembrane receptor for advanced glycation end products, which was first identified in 1992. It is a component of the immunoglobulin superfamily and is initially involved in immunological and chronic inflammatory reactions. It is believed that interaction between AGEs and RAGE on the endothelial cells contributes to the progression of peripheral neuropathy, since RAGE is expressed in the endothelial cells of peri- and endoneurial blood vessels. It was shown that when AGE binds to RAGE in endothelial cells, transcription factors NF-κB and AP-1 are activated. This results in the release of cytokines, including interleukin-6 and tumor necrosis factor, as well as vascular cell adhesion molecule-1. RAGE overexpression in endothelial cells in a diabetic transgenic mouse model demonstrated the significance of RAGE in the progression of diabetic neuropathy (DN). The progression of DN may be attributed to the synthesis and accumulation of AGEs in the peripheral nerve, which can directly or indirectly damage structural and functional proteins by activating AGE receptors. All the cell components in peripheral nerve tissues could be influenced by these pathologic events. Reduction of AGE levels by AGE breakers or antiglycation medications should be a potential therapeutic focus for the treatment of DN97, 98.
PURINOCEPTORS (P2Y, P2X)
In the purinergic complex, purine nucleotides such as ATP and ADP, along with the nucleoside adenosine, function as extracellular messengers, comprising a signaling mechanism. The existence of ATP receptors is suggested by purinergic signaling. A classification system was introduced in 1978 to distinguish adenosine receptors (P1) from ATP/ADP receptors (P2). P2 receptors are divided into two families based on their molecular structure and secondary messenger system: the P2X receptor, an ionotropic ligand-gated ion channel, and the P2Y receptor, a metabotropic G-protein-coupled receptor. To date, eight subclasses of P2Y receptors and seven subclasses of P2X receptors have been identified and studied in mammals. Except for P2Y12, P2Y13, and P2Y14, which couple to Gi proteins and inhibit adenylate cyclase, and P2Y11, which binds to Gs/Gq proteins, the majority of P2Y receptors bind to Gq/G11 proteins and thereby activate PLC-β. Both neurons and a majority of non-neuronal cells, including various pancreatic cells, exhibit functional purinoceptors. Complications from diabetes, such as painful diabetic neuropathy resulting in allodynia and hyperalgesia, have been observed. It has been demonstrated that sympathetic nerves in diabetic rats alter cutaneous polymodal receptors. Impaired healing following trauma and infection can be attributed to various factors, including edema, vascular complications, and peripheral neuropathy. It has been suggested that adenosine receptor agonists could accelerate the healing of cutaneous injuries, particularly those caused by diabetic foot ulcers. In some neuropathic pain models, P2 receptors or ATP/ADP receptors (P2X4 and P2Y12), and dorsal horn microglia are activated. Several studies have shown dramatically elevated levels of P2X2 and P2X3 receptor mRNA in the dorsal root ganglia of streptozotocin (STZ)-diabetic mice, indicating a possible correlation between the overexpression of these receptors and mechanical allodynia. Recent reports have highlighted a significant increase in P2X3 receptor expression and activity, which is believed to contribute to the development of chronic pain in STZ-induced diabetic rats. As an alternative strategy, the protective effects of adenosine were investigated using STZ-diabetic rat models of neuropathic pain. It was determined through the use of antagonists that the analgesic effects of adenosine were mediated by A1 receptors99, 100, 101, 102.
Nerve Growth Factor (NGF) Receptors
Histopathologically, diabetic neuropathy (DN) is characterized by abortive axonal regeneration and synaptogenesis, coupled with axonal degeneration, remyelination, demyelination, and atrophy. Experimental depletion of neurotrophic factors, their binding proteins, or their receptors can replicate many neural defects associated with diabetes. The survival of neurons, their resistance to apoptosis, and their capacity for rejuvenation depend on these neurotrophic substances. NGF and its related neurotrophin family peptides are among the most researched neurotrophic factors. Neurotrophins are thought to be vital in the diabetes-induced alterations in nerve structure and function, as they support the survival, preservation, and rejuvenation of neurons damaged by the disease. NGF interacts with two distinct types of receptors: the neurotrophin receptor p75 (p75NTR, a low-affinity receptor) and the tyrosine kinase receptors A: trkA, trkB, trkC (high-affinity receptors). Functionally, the trk receptors facilitate the binding of neurotrophins with high affinity, leading to signal transduction. Initially, p75 was identified as an NGF receptor with moderate affinity, believed to mainly participate in the formation of the functional NGF receptor and to modify the affinity of trkA for NGF binding. Further research has shown that p75 is extensively expressed in peripheral tissues, such as Schwann cells, and that its expression increases following nerve damage. Additionally, p75 has been found to modulate not only trkA but also trkB and trkC activities. NGF activity is specifically linked to p75 and/or trkA activation, which influences autonomic and small sensory nerve fibers. Conversely, trkC mediation of NT-3 signaling is essential for large nerve fibers, while the trkB high-affinity receptor mediates the effects of BDNF, NT-4, and NT-5 on medium-sized nerve fibers. It is important to note that p75 interacts with trk neurotrophin receptors to mediate neuronal survival during development, as demonstrated by transgenic studies through inhibiting apoptotic p75 signals. On the other hand, it has been shown that the autonomic ganglia of STZ-diabetic rats express lower levels of p75, resulting in decreased NGF signaling to the neurons. Regardless, one thing is clear: the low-affinity neurotrophin receptor is a critical target for therapeutic intervention in DN, as it is crucial for maintaining the integrity of small nerve fibers involved in pain, warm thermal perception, and sweating103, 104. The expression of the NGF receptors tropomyosin receptor kinase A (TrkA) and p75 neurotrophin receptor (p75NTR) is dynamically regulated in immune cells, suggesting a variable need for NGF depending on their state of differentiation and functional activity. NGF can have pro-inflammatory or anti-inflammatory effects. Studies of inflammatory and autoimmune diseases, marked by abnormal activation of immune cells and increased cytokine production, have revealed localized increases in NGF at inflammation sites. Initially, elevated NGF levels were found in the cerebrospinal fluid of multiple sclerosis patients, correlating closely with the disease course. Some research has shown that cytokines involved in inflammation, such as IL-β, TNF-α, and IL-6, promote NGF synthesis in various cell types. In inflammatory conditions, NGF levels rise due to its release by mast cells, macrophages, and lymphocytes. Upon binding to TrkA, the NGF-TrkA complex is internalized peripherally and transported to the cell soma, where it activates transcription factors affecting gene expression. Both in rodents and humans, cutaneous administration of NGF has been shown to lead to hyperalgesia within 1–3 hours. The rapid nociceptor sensitization of cutaneous receptors indicates that NGF plays a significant role in acute nociceptive responses and chronic pain. The peripheral and brain effects of anti-NGF in neuropathic pain remain unclear, and the efficacy of anti-NGF in reducing chronic pain through local administration in a rodent sciatic constriction injury (CCI) model still needs investigation by assessing NGF and substance P in the dorsal root ganglion (DRG) and spinal cord. Neuronal activation was measured using c-Fos in the anterior cingulate cortex and ventrolateral periaqueductal gray. At 14 days after CCI, anti-NGF induced a significant dose-dependent change in mechanical threshold, thermal withdrawal latency, and cold sensitivity, lasting for 5 hours. Anti-NGF treatment resulted in reduced NGF upregulation in the DRG and spinal cord post-CCI, while only increasing substance P levels in the DRG, which the treatment subsequently decreased. Anti-NGF caused a significant reduction of neuronal activation in the anterior cingulate cortex but not in the ventrolateral periaqueductal gray. This study provides the first evidence of the effects of anti-NGF on brain activity. Thus, our findings suggest that anti-NGF improves chronic neuropathic pain, acting directly on peripheral sensitization and indirectly on central sensitization. Anti-NGF antibodies represent a novel approach in pain management and have shown potential in treating certain pain conditions that are currently inadequately addressed. Tanezumab presents an exciting new class of analgesics with the capacity to revolutionize pain treatment105, 106, 107.
GLUTAMATE RECEPTORS
One of the main excitatory neurotransmitters within the mammalian central nervous system (CNS) is the glutamate receptor, which is involved in both normal cellular activity and cellular injury. Ionotropic glutamate receptors, glutamate-gated channels, were believed to be the only mechanism by which glutamate functioned in the mammalian brain until the mid-1980s. Later, it was discovered that some cellular processes in mammalian systems are influenced by a novel class of glutamate receptors known as metabotropic glutamate receptors (mGluRs), which are coupled to effector systems via GTP-binding proteins/G proteins. An explanation for the onset of neuropathy could be the presence of oxidative stress and malfunction in the mitochondria. mGluRs, specifically those in Group-II mGluRs, can halt this chain reaction of oxidative injury. The mGluR3 receptor can be directly or indirectly activated by inhibiting 'glutamate carboxypeptidase II (GCP-II)' to prevent glucose-induced damage to neurons & axons. In Dorsal Root Ganglion (DRG) neurons, the GCP-II inhibitor '2-(phosphonomethyl) pentanedioic acid (2-PMPA)' inhibits the damage that glucose causes to cells and neurite degeneration. The neuropeptidase GCP-II, previously known as NAALADase, is highly expressed in glial cells and hydrolyzes the neuropeptide 'N-Acetyl-Aspartyl-Glutamate (NAAG)' into NAA. NAAG functions as an agonist at mGluR3 to protect neurons. Despite the lack of evidence for NAAG action at mGluR2, the agonist APDC exhibits activity at both mGluR2 and mGluR3, suggesting that mGluR2 activation may also have neuroprotective effects.
Similarly, animal models of diabetic neuropathy (DN) have shown signs of neuroprotection. In inbred type-1 diabetic BB/Wor rats, inhibitors of GCP-II have been shown to have a positive effect on hyperalgesia, neurophysiological, and structural degenerative DN changes, most likely by elevating NAAG levels. Treatment with the direct mGluR3 agonist 'LY379268' appears to offer substantial protection against diabetic neuropathy, according to preliminary experiments. After administering streptozotocin to induce diabetes in rats and administering the mGluR3 agonist 'LY379268', electrophysiological methodologies were utilized to assess differences in neuropathy severity from the earliest stage. Based on nerve conduction studies, LY379268 reduces the induction of DN. In diabetic neurons and Schwann cells, mGluR3 appears to regulate mitochondrial function, similar to its effect in cell culture. TGF-β antagonism is another possible mechanism through which mGluR3 agonists can prevent diabetic neuropathy. TGF-β1 and TGF-β2 mRNA are upregulated in diabetic DRG rats, but not TGF-β3108, 109, 110, 111, 112.
CANNABINOID RECEPTORS
For centuries, people have utilized the bioactive constituents of Cannabis sativa extracts, such as hashish and marijuana, referred to as cannabinoids, to alleviate pain. However, the understanding of the mechanisms behind cannabinoid activity has only been gained in the last few decades, thanks to the advancement of new experimental methodologies. It has been shown that endogenous ligands for cannabinoid receptors exist. Both endogenous and exogenous cannabinoids primarily target the cannabinoid receptor-1 (CB-1) and cannabinoid receptor-2 (CB-2). The neurons of the central and peripheral nervous systems are the main cell types expressing CB-1 receptors, closely related to the processing and modulation of nociceptive signals in the laminae of the spinal cord dorsal horn. Dorsal root ganglia have also been shown to possess CB-1 receptors. Some non-neuronal tissues, specifically immune cells, express both central and peripheral CB-2 receptors. It has been observed that mast cell activity is down-regulated when CB-2 receptors are activated. There is evidence that rat brain microglia harbor CB-2 receptors as well. CB-2 receptors likely play a role in mediating the analgesic effects of endocannabinoids, especially in the presence of inflammation. A study also demonstrated that STZ-induced hyperalgesia was dose-dependently relieved by single or long-term administration of the non-selective cannabinoid agonist ‘WIN-55,212-2,’ as well as possibly selective CB-1 cannabinoid receptor agonist ‘Met-F-AEA’ and selective CB-2 cannabinoid receptor agonist ‘AM1241’113, 114, 115. Neural cells cultured in conditions similar to hyperglycemia and neurons derived from diabetic rats appear to show a down-regulation of CB-1 receptor protein. TRPV1 receptor signaling is thought to increase as a direct effect of a decrease in CB-1 expression. Diabetes-mediated induction of thermal hyperalgesia and allodynia in mice is attributed to over-regulation of TRPV1 receptors on sensory neurons. However, a study also revealed that anandamide treatment, resulting in glucose intolerance in rats, raises a potential caution when using CB-1 receptor agonists to prevent neurodegeneration in diabetes. This could be attributed to a decline in the pancreas's glucose-dependent insulin secretion, which may have implications for type-2 diabetes. On the other hand, if obese type-2 diabetics are prescribed the selective CB-1 antagonist ‘SR141716’ (rimonabant), it is also imperative to consider the potential adverse effects on the neurological system. An overall reduced CB-1 receptor expression in nerve cells is linked to high glucose levels; thus, CB-1 receptors are promising therapeutic targets in halting the neurodegenerative process in diabetes due to the neuroprotective effect of cannabis116, 117. Additionally, numerous studies have demonstrated that the endocannabinoid system employs endocannabinoid ligands like AEA and 2-AG to modulate pain perception. Peripherally located CB-2 receptors are involved in mediating analgesic effects but are not associated with psychoactive effects. In an evaluation of the CB-2 receptor agonist ‘AM1241,’ the impact on STZ-induced diabetic neuropathy (DN) was observed. Activation of CB-2 receptors was found to reduce cyclooxygenase (COX), nitric oxide synthases (NOSs), and the transmission of pain in neuropathic pain caused by STZ. A significant in vivo study has confirmed that the agonist AM1241, which activates CB-2, dose-dependently reduces experimental neuropathic pain without causing any adverse effects on the central nervous system. By decreasing mechanical allodynia in db/db mice, CB-2 agonists ‘JWH-015’ and ‘JWH-133’ showed a potent anti-nociceptive effect. The research also revealed that the agonistic activity of the CB-2 receptor is suitable for treating painful DN. In diabetic rats, the spinal administration of the CB-2 agonist ‘WIN-55,212–2’ exhibited a potent anti-nociceptive effect, improved nerve conduction velocity (NCV), and reduced necrosis of the sciatic nerve118, 119, 120, 121, 122.
SEROTONIN RECEPTORS
Neuropathy can affect every neuron and signaling pathway involved in pain perception. The descending inhibitory pain pathways of adrenergic and serotonergic neurons are two of the most important systems in the regulation of neuropathic pain. Serotonin, or 5-hydroxytryptamine (5-HT), is considered a key neurotransmitter involved in the regulation of nociceptive transmission in the brain and spinal cord. Its intricate function in the management of pain is noteworthy. Unremitting pain may be connected to the potentiation of the declining facilitatory serotonin pathway or a constriction of the declining inhibitory serotonin system. It has been suggested that serotonin 5-HT2A and 5-HT3 receptors contribute to the spinal antinociceptive action of 5-HT. Mechanical hyperalgesia produced by 5-HT is mediated by serotonin 5-HT2B receptors; serotonin 5-HT2A and 5-HT3 receptors are not involved. Diverse researchers have shown that serotonin directly causes hyperalgesia in the main afferent neuron through the subset of serotonin receptors 5-HT1A; antagonists of the ‘5-HT (2A/2C) & 5-HT3’ receptors didn't significantly lessen mechanical hyperalgesia123, 124, 125. In a study, it was found that the 5-HT3 receptor antagonist ‘ondansetron’ and the serotonin 5-HT (2A/2C) receptor antagonist ‘ketanserin’ were found to be effective in examining the association of the serotonergic descending inhibitory pathway in the antihyperalgesic effect of ‘zonisamide’. The findings of this study suggest that the antihyperalgesic effects of ‘zonisamide’ are caused by the inhibition of enhancement of the thermal threshold as well as the partial inhibition of mechanical threshold by serotonin 5-HT (2A/2C) and 5-HT3 receptors of the serotonergic descending inhibitory pain pathways. It is evident from these outcomes that serotonin 5-HT (2A/2C) and 5-HT3 receptors are not involved in mechanical hyperalgesia in diabetic neuropathy (DN) produced by STZ126.
ADRENERGIC RECEPTORS
It was found that certain selective norepinephrine reuptake medications provide a more significant analgesic effect in diabetic neuropathy than relatively selective serotonin reuptake inhibitors. An earlier study showed that pretreatment with yohimbine counteracted the outcome of norepinephrine injection, which resulted in more significant antinociception in STZ-induced diabetic mice than in non-diabetic mice. Later, another exploration established that the intrathecally administered 5-HT showed vastly reduced antinociceptive potency in diabetic mice compared to non-diabetic mice. These findings indicate that the spinal noradrenergic system, as opposed to the serotonergic system, is a more potent regulator of the analgesic effects in diabetes. In diabetic mice, the focus was on the spinal α-adrenoceptors. An α2-Adreno receptor agonist called ‘clonidine’, administered intrathecally, induced a strong analgesic effect in both diabetic and non-diabetic rats. Nonetheless, neither diabetic nor non-diabetic mice showed any analgesic effect from an intraperitoneal infusion of the α1-Adreno receptor agonist ‘methoxamine’. These outcomes indicate that, while the α1-Adreno receptor is unaffected, spinal cord α2-Adreno receptors are pivotal for the analgesic effects brought on by the activation of α2-Adreno receptor agonists. The study also concluded that the α1-Adreno receptor-mediated analgesic effect was not observed in mice with or without diabetes. It has been proven that diabetic mesenteric arteries have escalated contractile responses to α1-adreno receptors. Although spinal cord α1-adreno receptors have shown to have no involvement in antinociceptive activity in diabetic animals, they do play a significant part in vasoconstriction. The over-regulation of α2-Adreno receptors was produced by a decrease in norepinephrine release in the spinal cord of mice with diabetes, indicating that the antinociceptive effect was amplified by α2-Adreno receptor activation. Additionally, α2-Adreno receptors acted as a mediator of hypersensitivity. Therefore, neuropathic pain caused by diabetes is significantly moderated by the spinal noradrenergic systems. For the treatment of DN pain, selective norepinephrine reuptake inhibitors and α2-Adrenergic agonist-related activation of α2-adreno receptors may be useful.
There are several genes, ion channels, and enzymes that are either upregulated or downregulated in individuals with diabetic peripheral neuropathy (DPN). There is an upregulation of T-cell induced transcriptional activators: NFIL3 (Nuclear Factor Interleukin Regulated Protein 3), IFI16 (Interferon-gamma inducible protein 16), BCL6, & BCOR (transcription repressor & its co-repressor, respectively); the last two are indicative of inflammatory cell infiltration. Downregulation of transcriptional factor (enriched in nociceptors): ISL2, TLX3, PRDM12; neuronal transcriptional factors: NHLH1, SCRT1, SCRT2, SIX4, etc., almost half of the ion channels which are downregulated in DPN are K+ channels (K+ voltage-gated channels: KCNH2, KCNQ2) expressed in Aβ fibers. ASIC3 (acid-sensing ion channel 3), a prominent pain-transducing ion channel expressed in C-fibers, was downregulated. GLRA3 (glycine receptor), which contributes to PGE3-related pain hypersensitivity, is upregulated. Among GPCRs: inflammation-related pathways are upregulated as neuronal modulatory genes were downregulated in DPN. DPN subjects also show decreased expression of adenosine (ADORA1); norepinephrine (ADRA2C); dynorphin (OPRK1), all of which have antinociceptive properties. Downregulation of these receptors can either cause increased nociceptive hypersensitivity or neuronal loss. DPN individuals show downregulation of the NeuN gene (a well-established neuronal marker), and Synuclein Gamma (SNCG; highly expressed in the peripheral nervous system & shows decreased expression in DPN). A key enzyme, NAT8L, which catalyzes the formation of N-acetylaspartate (NAA) important for neuronal health, is downregulated127, 128, 129.
BRADYKININ (BK) RECEPTORS
Hyperalgesia and neuropathic pain are persistently present in cases of diabetic neuropathy. Moreover, hyperalgesia is a primary signal of diabetic neuropathy. It is a signal of early anomalies that precede the overt manifestation of DN, according to a large cohort study. In one study, sustained hyperglycemia developed 72 hours following treatment with STZ. Alongside STZ's diabetogenic effects, irreversible hyperalgesia also developed. The invention of the vincristine model of chemotherapy-induced painful toxic neuropathy has made it possible to study the mechanisms behind this type of neuropathic pain and track its progression. Researchers indicate that vincristine increases the quantity of unmyelinated sensory axons and causes the axonal microtubule cytoskeleton to become disorganized. Daily administration of vincristine led to a gradual reduction in pain threshold. Notably, hyperalgesia persisted even after cessation of the vincristine treatment, remaining conspicuous on day 24 of the trial. Unlike STZ, which caused persistent and long-lasting hyperalgesia, vincristine-induced hyperalgesia was reversible because the drug’s nociceptive threshold progressively returned to its original values upon withdrawal. BK is now known to be a neuromodulator and regulator of multiple vascular as well as organ functions in addition to being a proinflammatory mediator. BK is involved in the induction and regulation of pain stimuli’s transmission130, 131, 132.
There is a scarcity of data on BK's involvement in diabetic neuropathy. Rats with STZ-induced diabetes have both B1 and B2 receptors dispersed throughout their brains and spinal cords. In the year 2000, hypoalgesia in B1 receptor-deficient mice was reported133. In animal models of DN, it has been suggested that the kinin system mediates hyperalgesia through the inducible BK-B1 receptor subclass. When STZ was administered to B1 receptor knockout mice in the type-1 diabetes model, diabetic hyperalgesia was not present. The STZ-induced diabetic thermal hyperalgesia in mice is also reduced by long-term use of the selective BK-B1 receptor antagonists ‘R-715 and R954’. There is a dearth of evidence and literature about the involvement of B2 receptors in DN/hyperalgesia, in contrast to the significance of B1 receptors in DN pain mechanisms. Treatment with a specific B2 receptor antagonist, ‘HOE 140’, prevents vincristine hyperalgesia and STZ from developing. ‘Des-Arg10-HOE 140’, a selective B1 receptor antagonist, on the other hand, does not induce vincristine hyperalgesia; rather, it merely delays and reduces the decline in the nociceptive threshold following STZ. The B1-B2 receptors are significant in the expression & activation of chemically induced hyperalgesia. However, as the STZ hyperalgesia paradigm shows, B2 receptors play an even more significant role. The only effects of ‘des-Arg10-HOE140’ in blocking B1 receptors were significant delay in the onset of hyperalgesia and a moderate reduction in the degree of decline in the nociceptive threshold134, 135, 136.
CHEMOKINE RECEPTORS
A broad class of proteins known as chemokines, also referred to as chemotactic cytokines, are pivotal for regulating leukocyte migration. Several studies have demonstrated the significant roles chemokines play in the neurologic system in addition to orchestrating the immune response. For instance, in a number of animal models, the pathophysiology of neuropathic pain has been linked to excitatory chemokine signaling. In a subset of animal models, including HIV-1-induced neuropathy and opiate-induced hyperalgesia, some findings have linked the pathophysiology of neuropathic pain to the chemokine stromal cell-derived factor-1 (SDF-1) and its receptor CXCR4. Peripheral nerve samples from diabetic patients with progressive diabetic neuropathy displayed elevated CXCR4 receptor expression as determined by microarray analysis, indicating a similar role for CXCR4 receptors in PDN. Based on this evidence, a study was conducted to assess whether the pathophysiology of PDN could be significantly influenced by excitatory CXCR4/SDF-1 signaling in DRG neurons. A peripheral nerve microarray analysis of diabetic patients with advanced diabetic neuropathy revealed elevated CXCR4 expression, suggesting a role for CXCR4/SDF-1 signaling in the etiology of diabetic neuropathy in humans. In two animal models of type II diabetes, it was established that the selective CXCR4 antagonist AMD3100 reversed PDN, indicating that continuous CXCR4/SDF-1 signaling is necessary for PDN manifestation. Additionally, compared to normal mice, SDF-1 elevated Ca2+ influx in DRG neurons from diabetic mice, pointing to enhanced CXCR4 receptor activation in DRG neurons in the context of PDN. It has also been shown that SDF-1 facilitates the infiltration of inflammatory cells into the DRG. It was discovered that diabetic rats’ DRGs had increased levels of SDF-1 expression in their neurons and a significant influx of inflammatory cells, which may have significant implications for how pain mechanisms are produced in these conditions. All things considered, these findings highlight the significance of CXCR4/SDF-1 signaling in PDN pathophysiology, as it activates DRG neurons and attracts inflammatory cells into diabetic DRG137, 138, 139, 140, 141.
MUSCARINIC RECEPTORS
In diabetic rodents, antagonists of the muscarinic 1 sub-class receptor (M1R) also alleviate numerous neuropathy symptoms, and their efficacy in HIV-associated neuropathy and chemotherapy models suggests a neuroprotective and regenerative potential independent of the disease. A reduced density of small sensory nerve fibers in the skin and cornea indicates nerve damage, which is increasingly recognized as a primary sign of diabetic neuropathy. In diabetic patients undergoing simultaneous pancreas and kidney transplantation, improvements in other neuropathy symptoms either followed or preceded the recovery of small fiber density. A study was conducted to determine whether the ability of muscarinic antagonists to increase neurite outgrowth in vitro and enhance the density of corneal and epidermal nerve fibers in diabetic rodents translated to individuals with established diabetic neuropathy. The muscarinic antagonist oxybutynin was chosen for the study. Agents that bind the M1R subtype possess the unexpected neuritogenic property, which could be blocked by knocking out or disrupting M1R. Neuritogenesis may be activated by M1R antagonism through signaling via the ‘CaMKKβ/AMPK/PGC-1α pathway’, which improves mitochondrial function, and tubulin cytoskeleton modulation, which promotes axonal transport. The M1, M2, and M3 receptor subtype antagonist oxybutynin was found to be neuritogenic in the current study. Its peak efficiency and degree of response were comparable to those of the M1R-specific antagonist muscarinic toxin (MT-7). Increases in mitochondrial basal, maximum, and spare respiratory capacity were observed in conjunction with neurogenesis. Since oxybutynin is approved for clinical use, it offers the possibility to investigate whether enhancing neurite outgrowth in vitro translates into the effectiveness of rejuvenating epidermal fibers. This makes oxybutynin's ability to stimulate neurite outgrowth relevant. Overall, in line with the results obtained, muscarinic antagonists were found safe for long-term use and may be a groundbreaking therapeutic option for the management of diabetic neuropathy142, 143, 144, 145, 146, 147, 148.
CONCLUSION
DN involves a complex interplay of metabolic and inflammatory pathways, with various receptors playing critical roles. Understanding these receptors' roles in the pathogenesis of DN can support the development of targeted therapeutics to alleviate nerve damage and relieve symptoms in diabetic patients.
Abbreviations
2-PMPA - 2-(Phosphonomethyl) Pentanedioic Acid, ADORA1 - Adenosine A1 Receptor, ADRA2C - Alpha-2C Adrenergic Receptor, AGE - Advanced Glycation End-products, ASIC3 - Acid-Sensing Ion Channel 3, BB/Wor - BioBreeding Worcester Rat, BCL6 - B-Cell Lymphoma 6, BCOR - BCL6 Corepressor, BDNF - Brain-Derived Neurotrophic Factor, BK - Bradykinin, CaMKKβ - Calcium/Calmodulin-Dependent Protein Kinase Kinase Beta, CCI - Chronic Constriction Injury, CD36 - Cluster of Differentiation 36, CX3C - C-X3-C Motif Chemokine, CXCR4 - C-X-C Chemokine Receptor Type 4, CXC - C-X-C Motif Chemokine, DNP - Diabetic Neuropathy Pain, DN - Diabetic Neuropathy, DR - Dorsal Root, DRG - Dorsal Root Ganglion, GABA - Gamma-Aminobutyric Acid, GCP-II - Glutamate Carboxypeptidase II, GLRA3 - Glycine Receptor Alpha 3, GPCR - G Protein-Coupled Receptor, HbA1c - Hemoglobin A1c, HIV-1 - Human Immunodeficiency Virus 1, HOE 140 - Icatibant (B2 Receptor antagonist), IFI16 - Interferon-γ Inducible Protein 16, IL-β - Interleukin Beta, IL-6 - Interleukin 6, , IKK - Inhibitor of Nuclear Factor kappa-B Kinase, ISL2 - Insulin Gene Enhancer Protein ISL-2, KCNH2 - Potassium Voltage-Gated Channel Subfamily H Member 2, KCNQ2 - Potassium Voltage-Gated Channel Subfamily Q Member 2, MAPK - Mitogen-Activated Protein Kinase, M1R - Muscarinic 1 Receptor, MOR - Mu Opioid Receptor, MRZ2/576 - Specific Glycine-Site NMDA Receptor Antagonist (Experimental), MT-7 - Muscarinic Toxin 7, NAAG - N-Acetyl-Aspartyl-Glutamate, NAA - N-Acetylaspartate, NAT8L - N-Acetyltransferase 8-Lik, eNF-κB - Nuclear Factor kappa-light-chain-enhancer of activated B cells, NFIL3 - Nuclear Factor, Interleukin 3 Regulated, NHLH1 - Nescient Helix Loop Helix 1, NMDA - N-Methyl-D-Aspartate, OPRK1 - Opioid Receptor, Kappa 1, PDN - Painful Diabetic Neuropathy, PGC-1α - Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha, PRDM12 - PR Domain Containing 12, RAGE - Receptor for Advanced Glycation End-products, SDF-1 - Stromal Cell-Derived Factor 1, SCRT1 - Scribble Planar Cell Polarity Protein, SCRT2 - Scratch Family Transcriptional Repressor 2, SIX4 - Sine Oculis Homeobox Homolog 4, SNCG - Synuclein Gamma, SR-A - Scavenger Receptor Class A, STZ - Streptozotocin, TGF-β - Transforming Growth Factor Beta, TLX3 - T-Cell Leukemia Homeobox 3, TNF-α - Tumor Necrosis Factor Alpha, TRP - Transient Receptor Potential, TRPA1 - Transient Receptor Potential Ankyrin 1, TRPV1 - Transient Receptor Potential Vanilloid 1, TRPV4 - Transient Receptor Potential Vanilloid 4
Acknowledgments
The authors thank Integral University for providing an excellent research facility and manuscript communication number (IU/R&D/2024-MCN0002825).
Author’s contributions
Design & Conceptualization: Badruddeen; Outline & Data Collection: Garima Sharma, Deepa Neopane; Writing-Original draft, table & figure: Garima Sharma; Review & Editing: Juber Akhtar, Mohammad Irfan Khan, Mohammad Ahmad. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Kobayashi
M.,
Zochodne
D.W.,
Diabetic neuropathy and the sensory neuron: new aspects of pathogenesis and their treatment implications. Journal of Diabetes Investigation.
2018;
9
(6)
:
1239-54
.
View Article PubMed Google Scholar -
Sun
H.,
Saeedi
P.,
Karuranga
S.,
Pinkepank
M.,
Ogurtsova
K.,
Duncan
B.B.,
IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Research and Clinical Practice.
2022;
183
:
109119
.
View Article PubMed Google Scholar -
Pop-Busui
R.,
Boulton
A.J.,
Feldman
E.L.,
Bril
V.,
Freeman
R.,
Malik
R.A.,
Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care.
2017;
40
(1)
:
136-54
.
View Article PubMed Google Scholar -
Pop-Busui
R.,
Boulton
A.J.,
Feldman
E.L.,
Bril
V.,
Freeman
R.,
Malik
R.A.,
Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care.
2017;
40
(1)
:
136-54
.
View Article PubMed Google Scholar -
Waiz
M.,
Alvi
S.S.,
Ahmad
S.,
Khan
M.S.,
Association of PCSK-9 with the Biomarkers of Type-2 Diabetes and its Complications in the Indian Population: a Pilot Study. Clinical Laboratory.
2024;
70
(2)
.
View Article PubMed Google Scholar -
Jaiswal
M.,
Divers
J.,
Dabelea
D.,
Isom
S.,
Bell
R.A.,
Martin
C.L.,
Prevalence of and risk factors for diabetic peripheral neuropathy in youth with type 1 and type 2 diabetes: SEARCH for diabetes in youth study. Diabetes Care.
2017;
40
(9)
:
1226-32
.
View Article PubMed Google Scholar -
Mohammed
S.,
Mahmood
T.,
Shamim
A.,
Ahsan
F.,
Shariq
M.,
Parveen
S.,
Encyclopaedic Review of Glipizide Pre-clinical and Clinical Status. Drug Research (Stuttgart).
2024;
74
(3)
:
123-32
.
View Article PubMed Google Scholar -
Ray
P.,
Torck
A.,
Quigley
L.,
Wangzhou
A.,
Neiman
M.,
Rao
C.,
Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain.
2018;
159
(7)
:
1325-45
.
View Article PubMed Google Scholar -
Bhuiyan
S.A.,
Xu
M.,
Yang
L.,
Semizoglou
E.,
Bhatia
P.,
Pantaleo
K.I.,
Iv
undefined Robert W Gereau,
Harmonized cross-species cell atlases of trigeminal and dorsal root ganglia. Science Advances.
2024;
10
(25)
:
eadj9173
.
View Article PubMed Google Scholar -
Jung
M.,
Dourado
M.,
Maksymetz
J.,
Jacobson
A.,
Laufer
B.I.,
Baca
M.,
Cross-species transcriptomic atlas of dorsal root ganglia reveals species-specific programs for sensory function. Nature Communications.
2023;
14
(1)
:
366
.
View Article PubMed Google Scholar -
Jingxuan
L.,
Litian
M.,
Jianfang
F.,
Different drugs for the treatment of painful diabetic peripheral neuropathy: a meta-analysis. Frontiers in Neurology.
2021;
12
:
682244
.
View Article PubMed Google Scholar -
Feldman
E.L.,
Nave
K.A.,
Jensen
T.S.,
Bennett
D.L.,
New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron.
2017;
93
(6)
:
1296-313
.
View Article PubMed Google Scholar -
Cashman
C.R.,
Höke
A.,
Mechanisms of distal axonal degeneration in peripheral neuropathies. Neuroscience Letters.
2015;
596
:
33-50
.
View Article PubMed Google Scholar -
Lupachyk
S.,
Watcho
P.,
Stavniichuk
R.,
Shevalye
H.,
Obrosova
I.G.,
Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes.
2013;
62
(3)
:
944-52
.
View Article PubMed Google Scholar -
Ma
J.,
Pan
P.,
Anyika
M.,
Blagg
B.S.,
Dobrowsky
R.T.,
Modulating molecular chaperones improves mitochondrial bioenergetics and decreases the inflammatory transcriptome in diabetic sensory neurons. ACS Chemical Neuroscience.
2015;
6
(9)
:
1637-48
.
View Article PubMed Google Scholar -
Hashim
M.,
Badruddeen
undefined,
Akhtar
J.,
Khan
M.I.,
Ahmad
M.,
Islam
A.,
Diabetic Neuropathy: An Overview of Molecular Pathways and Protective Mechanisms of Phytobioactives. Endocrine, Metabolic & Immune Disorders Drug Targets.
2024;
24
(7)
:
758-76
.
View Article PubMed Google Scholar -
Kidwai
M.Z.,
Alam
R.,
Ahsan
H.,
Khan
M.M.,
Khan
S.,
Analysis of malondialdehyde and ferric reducing ability of plasma in type 2 diabetes mellitus patients in Lucknow city, India. Acta Biochimica Indonesiana.
2022;
5
(1)
:
84
.
View Article Google Scholar -
Hur
J.,
Dauch
J.R.,
Hinder
L.M.,
Hayes
J.M.,
Backus
C.,
Pennathur
S.,
The metabolic syndrome and microvascular complications in a murine model of type 2 diabetes. Diabetes.
2015;
64
(9)
:
3294-304
.
View Article PubMed Google Scholar -
Hur
J.,
O'Brien
P.D.,
Nair
V.,
Hinder
L.M.,
McGregor
B.A.,
Jagadish
H.V.,
Transcriptional networks of murine diabetic peripheral neuropathy and nephropathy: common and distinct gene expression patterns. Diabetologia.
2016;
59
(6)
:
1297-306
.
View Article PubMed Google Scholar -
Skapek
S.X.,
Ferrari
A.,
Gupta
A.A.,
Lupo
P.J.,
Butler
E.,
Shipley
J.,
Rhabdomyosarcoma. Nature Reviews. Disease Primers.
2019;
5
(1)
:
1-8
.
View Article PubMed Google Scholar -
Khan
M.D.,
Ahmad
M.K.,
Alam
R.,
Jaiswal
G.,
Khan
M.M.,
Utility of Indian diabetes risk score for the screening of type 2 diabetes mellitus and cardiovascular disease in and around areas of Lucknow. International Journal of Diabetes in Developing Countries.
2023;
43
(6)
:
892-8
.
View Article Google Scholar -
Kobayashi
M.,
Chandrasekhar
A.,
Cheng
C.,
Martinez
J.A.,
Ng
H.,
Hoz
C. de la,
Diabetic polyneuropathy, sensory neurons, nuclear structure and spliceosome alterations: a role for CWC22. Disease Models & Mechanisms.
2017;
10
(3)
:
215-24
.
View Article PubMed Google Scholar -
Thiel
G.,
Guethlein
L.A.,
Rössler
O.G.,
Insulin-Responsive Transcription Factors. Biomolecules.
2021;
11
(12)
:
1886
.
View Article PubMed Google Scholar -
A. Belfiore,
R. Malaguarnera,
V. Vella,
M.C. Lawrence,
L. Sciacca,
F. Frasca,
A. Morrione,
R. Vigneri,
Insulin receptor isoforms in physiology and disease: an updated view. Endocrine reviews.
2017;
38
(5)
:
379-431
.
View Article Google Scholar -
Glasgow
N.G.,
Siegler Retchless
B.,
Johnson
J.W.,
Molecular bases of NMDA receptor subtype-dependent properties. The Journal of Physiology.
2015;
593
(1)
:
83-95
.
View Article PubMed Google Scholar -
Lambert
D.G.,
Opioids and opioid receptors; understanding pharmacological mechanisms as a key to therapeutic advances and mitigation of the misuse crisis. BJA open.
2023;
6
:
100141
.
View Article Google Scholar -
McDonald
J.,
Lambert
D.G.,
Opioid receptors. BJA Education.
2015;
15
(5)
:
219-224
.
View Article Google Scholar -
Stein
C.,
Opioid Receptors. Annual review of medicine.
2016;
67
(1)
:
433-51
.
View Article PubMed Google Scholar -
Toll
L.,
Bruchas
M.R.,
Calo
G.,
Cox
B.M.,
Zaveri
N.T.,
Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacological reviews.
2016;
68
(2)
:
419-57
.
View Article Google Scholar -
Sobczak
M.,
Sałaga
M.,
Storr
M.A.,
Fichna
J.,
Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: current concepts and future perspectives. Journal of Gastroenterology.
2014;
49
(1)
:
24-45
.
View Article PubMed Google Scholar -
Ramasamy
R.,
Yan
S.F.,
Schmidt
A.M.,
Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Annals of the New York Academy of Sciences.
2011;
1243
(1)
:
88-102
.
View Article PubMed Google Scholar -
Lee
E.J.,
Park
J.H.,
Receptor for Advanced Glycation Endproducts (RAGE), Its Ligands, and Soluble RAGE: Potential Biomarkers for Diagnosis and Therapeutic Targets for Human Renal Diseases. Genomics & Informatics.
2013;
11
(4)
:
224-9
.
View Article PubMed Google Scholar -
Yue
Q.,
Song
Y.,
Liu
Z.,
Zhang
L.,
Yang
L.,
Li
J.,
Receptor for Advanced Glycation End Products (RAGE): A Pivotal Hub in Immune Diseases. Molecules (Basel, Switzerland).
2022;
27
(15)
:
4922
.
View Article PubMed Google Scholar -
Dong
H.,
Zhang
Y.,
Huang
Y.,
Deng
H.,
Pathophysiology of RAGE in inflammatory diseases. Frontiers in Immunology.
2022;
13
:
931473
.
View Article PubMed Google Scholar -
Burnstock
G.,
Purine and purinergic receptors. Brain and Neuroscience Advances.
2018;
2
:
2398212818817494
.
View Article PubMed Google Scholar -
Díaz-Muñoz
M.,
Hernández-Muñoz
R.,
Butanda-Ochoa
A.,
Structure-activity features of purines and their receptors: implications in cell physiopathology. Molecular Biomedicine.
2022;
3
(1)
:
5
.
View Article PubMed Google Scholar -
Catchen
M.J.,
John
H.,
The zebrafish: genetics, genomics and informatics. Academic Press; 2011 Sep 14. 2011.
Google Scholar -
Aloe
L.,
Rocco
M.L.,
Balzamino
B.O.,
Micera
A.,
Nerve Growth Factor: A Focus on Neuroscience and Therapy. Current Neuropharmacology.
2015;
13
(3)
:
294-303
.
View Article PubMed Google Scholar -
Rocco
M.L.,
Soligo
M.,
Manni
L.,
Aloe
L.,
Nerve Growth Factor: Early Studies and Recent Clinical Trials. Current neuropharmacology.
2018;
16
(10)
:
1455-65
.
View Article Google Scholar -
Hansen
K.B.,
Wollmuth
L.P.,
Bowie
D.,
Furukawa
H.,
Menniti
F.S.,
Sobolevsky
A.I.,
Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacological Reviews.
2021;
73
(4)
:
298-487
.
View Article PubMed Google Scholar -
Brassai
A.,
Suvanjeiev
R.G.,
Bán
E.G.,
Lakatos
M.,
Role of synaptic and nonsynaptic glutamate receptors in ischaemia induced neurotoxicity. Brain Research Bulletin.
2015;
112
:
1-6
.
View Article PubMed Google Scholar -
Petralia
R.S.,
Rubio
M.E.,
Wang
Y.X.,
Wenthold
R.J.,
Differential distribution of glutamate receptors in the cochlear nuclei. Hearing Research.
2000;
147
(1-2)
:
59-69
.
View Article PubMed Google Scholar -
Shahbazi
F.,
Grandi
V.,
Banerjee
A.,
Trant
J.F.,
Cannabinoids and Cannabinoid Receptors: The Story so Far. iScience.
2020;
23
(7)
:
101301
.
View Article PubMed Google Scholar -
Laprairie
R.B.,
Bagher
A.M.,
Kelly
M.E.,
Denovan-Wright
E.M.,
Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. British Journal of Pharmacology.
2015;
172
(20)
:
4790-805
.
View Article PubMed Google Scholar -
Haspula
D.,
Clark
M.A.,
Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. International Journal of Molecular Sciences.
2020;
21
(20)
:
7693
.
View Article PubMed Google Scholar -
Zweckstetter
M.,
Dityatev
A.,
Ponimaskin
E.,
Structure of serotonin receptors: molecular underpinning of receptor activation and modulation. Signal Transduction and Targeted Therapy.
2021;
6
(1)
:
243
.
View Article PubMed Google Scholar -
Kanova
M.,
Kohout
P.,
Serotonin-Its Synthesis and Roles in the Healthy and the Critically Ill. International Journal of Molecular Sciences.
2021;
22
(9)
:
4837
.
View Article PubMed Google Scholar -
Kuhar
M.J.,
Couceyro
P.R.,
Lambert
P.D.,
α- and β-Adrenergic Receptors. In: Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999. 1999
.
-
Graham
R.M.,
Adrenergic receptors: structure and function. Cleveland Clinic Journal of Medicine.
1990;
57
(5)
:
481-91
.
View Article PubMed Google Scholar -
García-Sáinz
J.A.,
Romero-Ávila
M.T.,
Medina
L.C.,
α(1D)-Adrenergic receptors constitutive activity and reduced expression at the plasma membrane. Methods in Enzymology.
2010;
484
:
109-25
.
View Article PubMed Google Scholar -
Taylor
B.N.,
Cassagnol
M.,
Alpha-Adrenergic Receptors. [Updated 2023 Jul 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov /books/NBK539830. 2024
.
-
Shen
J.K.,
Zhang
H.T.,
Function and structure of bradykinin receptor 2 for drug discovery. Acta Pharmacologica Sinica.
2023;
44
(3)
:
489-98
.
View Article PubMed Google Scholar -
Pirahanchi
Y.,
Sharma
S.,
Physiology, Bradykinin. [Updated 2023 Jul 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537187/. 2024
.
-
Marceau
F.,
Bachelard
H.,
Bouthillier
J.,
Fortin
J.P.,
Morissette
G.,
Bawolak
M.T.,
Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. International Immunopharmacology.
2020;
82
:
106305
.
View Article PubMed Google Scholar -
Hughes
C.E.,
Nibbs
R.J.,
A guide to chemokines and their receptors. The FEBS Journal.
2018;
285
(16)
:
2944-71
.
View Article PubMed Google Scholar -
Kufareva
I.,
Gustavsson
M.,
Zheng
Y.,
Stephens
B.S.,
Handel
T.M.,
What Do Structures Tell Us About Chemokine Receptor Function and Antagonism?. Annual Review of Biophysics.
2017;
46
(1)
:
175-98
.
View Article PubMed Google Scholar -
Gustavsson
M.,
New insights into the structure and function of chemokine receptor:chemokine complexes from an experimental perspective. Journal of Leukocyte Biology.
2020;
107
(6)
:
1115-22
.
View Article PubMed Google Scholar -
Saternos
H.C.,
Almarghalani
D.A.,
Gibson
H.M.,
Meqdad
M.A.,
Antypas
R.B.,
Lingireddy
A.,
Distribution and function of the muscarinic receptor subtypes in the cardiovascular system. Physiological Genomics.
2018;
50
(1)
:
1-9
.
View Article PubMed Google Scholar -
Abrams
P.,
Andersson
K.E.,
Buccafusco
J.J.,
Chapple
C.,
de Groat
W.C.,
Fryer
A.D.,
Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. British Journal of Pharmacology.
2006;
148
(5)
:
565-78
.
View Article PubMed Google Scholar -
Uhlén
M.,
Fagerberg
L.,
Hallström
B.M.,
Lindskog
C.,
Oksvold
P.,
Mardinoglu
A.,
Proteomics. Tissue-based map of the human proteome. Science.
2015;
347
(6220)
:
1260419
.
View Article PubMed Google Scholar -
Samanta
A.,
Hughes
T.E.,
Moiseenkova-Bell
V.Y.,
Transient Receptor Potential (TRP) Channels. Sub-Cellular Biochemistry.
2018;
87
:
141-65
.
View Article PubMed Google Scholar -
G. Vazquez,
B.J. Wedel,
O. Aziz,
M. Trebak,
Jr. J.W. Putney,
The mammalian TRPC cation channels. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research.
2004;
1742
(1-3)
:
21-36
.
View Article PubMed Google Scholar -
Vazquez
G.,
Wedel
B.J.,
Aziz
O.,
Trebak
M.,
Putney
J.W.,
The enigmatic TRPCs: multifunctional cation channels. Trends in cell biology.
2004;
14
(6)
:
282-6
.
View Article Google Scholar -
Wes
P.D.,
Chevesich
J.,
Jeromin
A.,
Rosenberg
C.,
Stetten
G.,
Montell
C.,
TRPC1, a human homolog of a Drosophila store-operated channel. Proceedings of the National Academy of Sciences.
1995;
92
(21)
:
9652-6
.
View Article Google Scholar -
X.P. Dong,
X. Wang,
H. Xu,
TRP channels of intracellular membranes. Journal of neurochemistry.
2010;
113
(2)
:
313-28
.
View Article PubMed Google Scholar -
Zheng
J.,
Molecular mechanism of TRP channels. Comprehensive Physiology.
2013;
3
(1)
:
221-42
.
View Article PubMed Google Scholar -
Sameer
A.S.,
Nissar
S.,
Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. BioMed Research International.
2021;
2021
:
1157023
.
View Article PubMed Google Scholar -
Brennan
J.J.,
Gilmore
T.D.,
Evolutionary origins of tolllike receptor signaling. Molecular Biology and Evolution.
2018;
35
(7)
:
1576-87
.
View Article PubMed Google Scholar -
Leulier
F.,
Lemaitre
B.,
Toll-like receptors- taking an evolutionary approach. Nature Reviews. Genetics.
2008;
9
(3)
:
165-78
.
View Article PubMed Google Scholar -
El-Zayat
S.R.,
Sibaii
H.,
Mannaa
F.A.,
Toll-like receptors activation, signaling and targeting: an overview. Bulletin of the National Research Center.
2019;
43
(1)
:
187
.
View Article Google Scholar -
Zhang
X.,
Zhu
X.,
Bi
X.,
Huang
J.,
Zhou
L.,
The Insulin Receptor: An Important Target for the Development of Novel Medicines and Pesticides. International Journal of Molecular Sciences.
2022;
23
(14)
:
7793
.
View Article PubMed Google Scholar -
Stone
T.W.,
Subtypes of NMDA receptors. General Pharmacology.
1993;
24
(4)
:
825-32
.
View Article PubMed Google Scholar -
Belfiore
A.,
Malaguarnera
R.,
Vella
V.,
Lawrence
M.C.,
Sciacca
L.,
Frasca
F.,
Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocrine Reviews.
2017;
38
(5)
:
379-431
.
View Article PubMed Google Scholar -
Basso
L.,
Altier
C.,
Transient receptor potential channels in neuropathic pain. Current Opinion in Pharmacology.
2017;
32
:
9-15
.
View Article PubMed Google Scholar -
Cui
Y.Y.,
Xu
H.,
Wu
H.H.,
Qi
J.,
Shi
J.,
Li
Y.Q.,
Spatio-temporal expression and functional involvement of transient receptor potential vanilloid 1 in diabetic mechanical allodynia in rats. PLoS One.
2014;
9
(7)
:
e102052
.
View Article PubMed Google Scholar -
Ristoiu
V.,
Shibasaki
K.,
Uchida
K.,
Zhou
Y.,
Ton
B.T.,
Flonta
M.L.,
Hypoxia-induced sensitization of transient receptor potential vanilloid 1 involves activation of hypoxia-inducible factor-1 alpha and PKC. Pain.
2011;
152
(4)
:
936-45
.
View Article PubMed Google Scholar -
Narayanaswamy
H.,
Facer
P.,
Misra
V.P.,
Timmers
M.,
Byttebier
G.,
Meert
T.,
A longitudinal study of sensory biomarkers of progression in patients with diabetic peripheral neuropathy using skin biopsies. Journal of Clinical Neuroscience.
2012;
19
(11)
:
1490-6
.
View Article PubMed Google Scholar -
Khomula
E.V.,
Viatchenko-Karpinski
V.Y.,
Borisyuk
A.L.,
Duzhyy
D.E.,
Belan
P.V.,
Voitenko
N.V.,
Specific functioning of Cav3.2 T-type calcium and TRPV1 channels under different types of STZ-diabetic neuropathy. Biochimica et Biophysica Acta.
2013;
1832
(5)
:
636-49
.
View Article PubMed Google Scholar -
Koivisto
A.,
Hukkanen
M.,
Saarnilehto
M.,
Chapman
H.,
Kuokkanen
K.,
Wei
H.,
Inhibiting TRPA1 ion channel reduces loss of cutaneous nerve fiber function in diabetic animals: sustained activation of the TRPA1 channel contributes to the pathogenesis of peripheral diabetic neuropathy. Pharmacological Research.
2012;
65
(1)
:
149-58
.
View Article PubMed Google Scholar -
Eberhardt
M.J.,
Filipovic
M.R.,
Leffler
A.,
de la Roche
J.,
Kistner
K.,
Fischer
M.J.,
Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. The Journal of Biological Chemistry.
2012;
287
(34)
:
28291-306
.
View Article PubMed Google Scholar -
Andersson
D.A.,
Gentry
C.,
Light
E.,
Vastani
N.,
Vallortigara
J.,
Bierhaus
A.,
Methylglyoxal evokes pain by stimulating TRPA1. PLoS One.
2013;
8
(10)
:
e77986
.
View Article PubMed Google Scholar -
Huang
Q.,
Chen
Y.,
Gong
N.,
Wang
Y.X.,
Methylglyoxal mediates streptozotocin-induced diabetic neuropathic pain via activation of the peripheral TRPA1 and Nav1.8 channels. Metabolism: Clinical and Experimental.
2016;
65
(4)
:
463-74
.
View Article PubMed Google Scholar -
Zhu
T.,
Meng
Q.,
Ji
J.,
Lou
X.,
Zhang
L.,
Toll-like receptor 4 and tumor necrosis factor-alpha as diagnostic biomarkers for diabetic peripheral neuropathy. Neuroscience Letters.
2015;
585
:
28-32
.
View Article PubMed Google Scholar -
Hanke
M.L.,
Kielian
T.,
Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clinical Science (London, England).
2011;
121
(9)
:
367-87
.
View Article PubMed Google Scholar -
Pino
S.C.,
Kruger
A.J.,
Bortell
R.,
The role of innate immune pathways in type 1 diabetes pathogenesis. Current Opinion in Endocrinology, Diabetes, and Obesity.
2010;
17
(2)
:
126-30
.
View Article PubMed Google Scholar -
Dasu
M.R.,
Devaraj
S.,
Park
S.,
Jialal
I.,
Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care.
2010;
33
(4)
:
861-8
.
View Article PubMed Google Scholar -
Devaraj
S.,
Tobias
P.,
Jialal
I.,
Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine.
2011;
55
(3)
:
441-5
.
View Article PubMed Google Scholar -
Yamakawa
I.,
Kojima
H.,
Terashima
T.,
Katagi
M.,
Oi
J.,
Urabe
H.,
Inactivation of TNF-α ameliorates diabetic neuropathy in mice. American Journal of Physiology. Endocrinology and Metabolism.
2011;
301
(5)
:
844-52
.
View Article PubMed Google Scholar -
Zenker
J.,
Ziegler
D.,
Chrast
R.,
Novel pathogenic pathways in diabetic neuropathy. Trends in Neurosciences.
2013;
36
(8)
:
439-49
.
View Article PubMed Google Scholar -
Singh
B.,
Xu
Y.,
McLaughlin
T.,
Singh
V.,
Martinez
J.A.,
Krishnan
A.,
Resistance to trophic neurite outgrowth of sensory neurons exposed to insulin. Journal of Neurochemistry.
2012;
121
(2)
:
263-76
.
View Article PubMed Google Scholar -
Kim
B.,
McLean
L.L.,
Philip
S.S.,
Feldman
E.L.,
Hyperinsulinemia induces insulin resistance in dorsal root ganglion neurons. Endocrinology.
2011;
152
(10)
:
3638-47
.
View Article PubMed Google Scholar -
Goswami
K.,
Badruddeen
undefined,
Arif
M.,
Akhtar
J.,
Khan
M.I.,
Ahmad
M.,
Flavonoids, Isoflavonoids and others Bioactives for Insulin Sensitizations. Current Diabetes Reviews.
2024;
20
(2)
:
e270423216247
.
View Article PubMed Google Scholar -
Zhou
H.Y.,
Chen
S.R.,
Pan
H.L.,
Targeting N-methyl-D-aspartate receptors for treatment of neuropathic pain. Expert Review of Clinical Pharmacology.
2011;
4
(3)
:
379-88
.
View Article PubMed Google Scholar -
Bai
H.P.,
Liu
P.,
Wu
Y.M.,
Guo
W.Y.,
Guo
Y.X.,
Wang
X.L.,
Activation of spinal GABAB receptors normalizes N-methyl-D-aspartate receptor in diabetic neuropathy. Journal of the Neurological Sciences.
2014;
341
(1-2)
:
68-72
.
View Article PubMed Google Scholar -
Martínez-Navarro
M.,
Maldonado
R.,
Baños
J.E.,
Why mu-opioid agonists have less analgesic efficacy in neuropathic pain?. European Journal of Pain (London, England).
2019;
23
(3)
:
435-54
.
View Article PubMed Google Scholar -
Balogh
M.,
Zádor
F.,
Zádori
Z.S.,
Shaqura
M.,
Király
K.,
Mohammadzadeh
A.,
Efficacy-based perspective to overcome reduced opioid analgesia of advanced painful diabetic neuropathy in rats. Frontiers in Pharmacology.
2019;
10
:
347
.
View Article PubMed Google Scholar -
Wada
R.,
Yagihashi
S.,
Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Annals of the New York Academy of Sciences.
2005;
1043
(1)
:
598-604
.
View Article PubMed Google Scholar -
Indyk
D.,
Bronowicka-Szyde\lko
A.,
Gamian
A.,
Kuzan
A.,
Advanced glycation end products and their receptors in serum of patients with type 2 diabetes. Scientific Reports.
2021;
11
(1)
:
13264
.
View Article PubMed Google Scholar -
Sanajou
D.,
Ghorbani Haghjo
A.,
Argani
H.,
Aslani
S.,
AGE-RAGE axis blockade in diabetic nephropathy: current status and future directions. European Journal of Pharmacology.
2018;
833
:
158-64
.
View Article PubMed Google Scholar -
Tsuda
M.,
Tozaki-Saitoh
H.,
Inoue
K.,
Pain and purinergic signaling. Brain Research. Brain Research Reviews.
2010;
63
(1-2)
:
222-32
.
View Article PubMed Google Scholar -
Xu
G.Y.,
Li
G.,
Liu
N.,
Huang
L.Y.,
Mechanisms underlying purinergic P2X3 receptor-mediated mechanical allodynia induced in diabetic rats. Molecular Pain.
2011;
7
:
60
.
View Article PubMed Google Scholar -
Burnstock
G.,
Novak
I.,
Purinergic signalling and diabetes. Purinergic Signalling.
2013;
9
(3)
:
307-24
.
View Article PubMed Google Scholar -
Kim
H.C.,
Cho
Y.J.,
Ahn
C.W.,
Park
K.S.,
Kim
J.C.,
Nam
J.S.,
Nerve growth factor and expression of its receptors in patients with diabetic neuropathy. Diabetic Medicine.
2009;
26
(12)
:
1228-34
.
View Article PubMed Google Scholar -
Pittenger
G.,
Vinik
A.,
Nerve growth factor and diabetic neuropathy. Experimental Diabesity Research.
2003;
4
(4)
:
271-85
.
PubMed Google Scholar -
da Silva
J.T.,
Evangelista
B.G.,
Venega
R.A.,
Seminowicz
D.A.,
Chacur
M.,
Anti-NGF treatment can reduce chronic neuropathic pain by changing peripheral mediators and brain activity in rats. Behavioural Pharmacology.
2019;
30
(1)
:
79-88
.
View Article PubMed Google Scholar -
Schmidt
R.E.,
Dorsey
D.A.,
Roth
K.A.,
Parvin
C.A.,
Hounsom
L.,
Tomlinson
D.R.,
Effect of streptozotocin-induced diabetes on NGF, P75(NTR) and TrkA content of prevertebral and paravertebral rat sympathetic ganglia. Brain Research.
2000;
867
(1-2)
:
149-56
.
View Article PubMed Google Scholar -
Patel
M.K.,
Kaye
A.D.,
Urman
R.D.,
Tanezumab: therapy targeting nerve growth factor in pain pathogenesis. Journal of Anaesthesiology, Clinical Pharmacology.
2018;
34
(1)
:
111-6
.
View Article PubMed Google Scholar -
Berent-Spillson
A.,
Robinson
A.M.,
Golovoy
D.,
Slusher
B.,
Rojas
C.,
Russell
J.W.,
Protection against glucose-induced neuronal death by NAAG and GCP II inhibition is regulated by mGluR3. Journal of Neurochemistry.
2004;
89
(1)
:
90-9
.
View Article PubMed Google Scholar -
Spillson
A.B.,
Russell
J.W.,
Metabotropic glutamate receptor regulation of neuronal cell death. Experimental Neurology.
2003;
184
:
97-105
.
View Article PubMed Google Scholar -
Thomas
A.G.,
Corse
A.M.,
Coccia
C.F.,
Bilak
M.M.,
Rothstein
J.D.,
Slusher
B.S.,
NAALADase inhibition protects motor neurons against chronic glutamate toxicity. European Journal of Pharmacology.
2003;
471
(3)
:
177-84
.
View Article PubMed Google Scholar -
Zhang
W.,
Slusher
B.,
Murakawa
Y.,
Wozniak
K.M.,
Tsukamoto
T.,
Jackson
P.F.,
GCPII (NAALADase) inhibition prevents long-term diabetic neuropathy in type 1 diabetic BB/Wor rats. Journal of the Neurological Sciences.
2002;
194
(1)
:
21-8
.
View Article PubMed Google Scholar -
Anjaneyulu
M.,
Berent-Spillson
A.,
Russell
J.W.,
Metabotropic glutamate receptors (mGluRs) and diabetic neuropathy. Current Drug Targets.
2008;
9
(1)
:
85-93
.
View Article PubMed Google Scholar -
Azad
S.C.,
Rammes
G.,
Cannabinoids in anaesthesia and pain therapy. Current Opinion in Anaesthesiology.
2005;
18
(4)
:
424-7
.
View Article PubMed Google Scholar -
Goya
P.,
Jagerovic
N.,
Hernandez-Folgado
L.,
Martín
M.I.,
Cannabinoids and neuropathic pain. Mini-Reviews in Medicinal Chemistry.
2003;
3
(7)
:
765-72
.
View Article PubMed Google Scholar -
Bujalska
M.,
Effect of cannabinoid receptor agonists on streptozotocin-induced hyperalgesia in diabetic neuropathy. Pharmacology.
2008;
82
(3)
:
193-200
.
View Article PubMed Google Scholar -
Duarte
J.M.,
Nogueira
C.,
Mackie
K.,
Oliveira
C.R.,
Cunha
R.A.,
Köfalvi
A.,
Increase of cannabinoid CB1 receptor density in the hippocampus of streptozotocin-induced diabetic rats. Experimental Neurology.
2007;
204
(1)
:
479-84
.
View Article PubMed Google Scholar -
Zhang
F.,
Hong
S.,
Stone
V.,
Smith
P.J.,
Expression of cannabinoid CB1 receptors in models of diabetic neuropathy. The Journal of Pharmacology and Experimental Therapeutics.
2007;
323
(2)
:
508-15
.
View Article PubMed Google Scholar -
Bayram
E.H.,
Sezer
A.D.,
Elçioğlu
H.K.,
Diabetic neuropathy and treatment strategy-new challenges and applications. Smart Drug Delivery System.
2016;
2016
:
373-388
.
View Article Google Scholar -
Javed
H.,
Azimullah
S.,
Haque
M.E.,
Ojha
S.K.,
Cannabinoid type 2 (CB2) receptors activation protects against oxidative stress and neuroinflammation associated dopaminergic neurodegeneration in rotenone model of Parkinson's disease. Frontiers in Neuroscience.
2016;
10
:
321
.
View Article PubMed Google Scholar -
Woodhams
S.G.,
Sagar
D.R.,
Burston
J.J.,
Chapman
V.,
The role of the endocannabinoid system in pain. Pain control.
2015;
227
:
119-43
.
View Article Google Scholar -
McDonnell
C.,
Leánez
S.,
Pol
O.,
The inhibitory effects of cobalt protoporphyrin IX and cannabinoid 2 receptor agonists in type 2 diabetic mice. International Journal of Molecular Sciences.
2017;
18
(11)
:
2268
.
View Article PubMed Google Scholar -
Kumawat
V.S.,
Kaur
G.,
Therapeutic potential of cannabinoid receptor 2 in the treatment of diabetes mellitus and its complications. European Journal of Pharmacology.
2019;
862
:
172628
.
View Article PubMed Google Scholar -
Saadé
N.E.,
Jabbur
S.J.,
Nociceptive behavior in animal models for peripheral neuropathy: spinal and supraspinal mechanisms. Progress in Neurobiology.
2008;
86
(1)
:
22-47
.
View Article PubMed Google Scholar -
Tanabe
M.,
Sakaue
A.,
Takasu
K.,
Honda
M.,
Ono
H.,
Centrally mediated antihyperalgesic and antiallodynic effects of zonisamide following partial nerve injury in the mouse. Naunyn-Schmiedeberg's archives of pharmacology.
2005;
372
(2)
:
107-14
.
View Article PubMed Google Scholar -
Lopes
L.S.,
Pereira
S.S.,
Silva
L.L.,
Figueiredo
K.A.,
Moura
B.A.,
Almeida
F.R.,
Antinociceptive effect of topiramate in models of acute pain and diabetic neuropathy in rodents. Life Sciences.
2009;
84
(3-4)
:
105-10
.
View Article PubMed Google Scholar -
Bektas
N.,
Arslan
R.,
Ozturk
Y.,
Zonisamide: antihyperalgesic efficacy, the role of serotonergic receptors on efficacy in a rat model for painful diabetic neuropathy. Life Sciences.
2014;
95
(1)
:
9-13
.
View Article PubMed Google Scholar -
Omiya
Y.,
Suzuki
Y.,
Yuzurihara
M.,
Murata
M.,
Aburada
M.,
Kase
Y.,
Antinociceptive effect of shakuyakukanzoto, a Kampo medicine, in diabetic mice. Journal of Pharmacological Sciences.
2005;
99
(4)
:
373-80
.
View Article PubMed Google Scholar -
Hall
B.E.,
Macdonald
E.,
Cassidy
M.,
Yun
S.,
Sapio
M.R.,
Ray
P.,
Transcriptomic analysis of human sensory neurons in painful diabetic neuropathy reveals inflammation and neuronal loss. Scientific Reports.
2022;
12
(1)
:
4729
.
View Article PubMed Google Scholar -
Omiya
Y.,
Yuzurihara
M.,
Suzuki
Y.,
Kase
Y.,
Kono
T.,
Role of α2-adrenoceptors in enhancement of antinociceptive effect in diabetic mice. European Journal of Pharmacology.
2008;
592
(1-3)
:
62-6
.
View Article PubMed Google Scholar -
Bujalska
M.,
Tatarkiewicz
J.,
Gumu\lka
S.W.,
Effect of bradykinin receptor antagonists on vincristine- and streptozotocin-induced hyperalgesia in a rat model of chemotherapy-induced and diabetic neuropathy. Pharmacology.
2008;
81
(2)
:
158-63
.
View Article PubMed Google Scholar -
Tang
H.B.,
Inoue
A.,
Iwasa
M.,
Hide
I.,
Nakata
Y.,
Substance P release evoked by capsaicin or potassium from rat cultured dorsal root ganglion neurons is conversely modulated with bradykinin. Journal of Neurochemistry.
2006;
97
(5)
:
1412-8
.
View Article PubMed Google Scholar -
Campos
M.M.,
Ongali
B.,
Thibault
G.,
Neugebauer
W.,
Couture
R.,
Autoradiographic distribution and alterations of kinin B(2) receptors in the brain and spinal cord of streptozotocin-diabetic rats. Synapse (New York, N.Y.).
2005;
58
(3)
:
184-92
.
View Article PubMed Google Scholar -
Pesquero
J.B.,
Araujo
R.C.,
Heppenstall
P.A.,
Stucky
C.L.,
Silva
J.A.,
Walther
T.,
Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proceedings of the National Academy of Sciences of the United States of America.
2000;
97
(14)
:
8140-5
.
View Article PubMed Google Scholar -
Gabra
B.H.,
Berthiaume
N.,
Sirois
P.,
Nantel
F.,
Battistini
B.,
The kinin system mediates hyperalgesia through the inducible bradykinin B1 receptor subtype: evidence in various experimental animal models of type 1 and type 2 diabetic neuropathy. Biological Chemistry.
2006;
387
(2)
:
127-143
.
View Article Google Scholar -
Gabra
B.H.,
Benrezzak
O.,
Pheng
L.H.,
Duta
D.,
Daull
P.,
Sirois
P.,
Inhibition of type 1 diabetic hyperalgesia in streptozotocin-induced Wistar versus spontaneous gene-prone BB/Worchester rats: efficacy of a selective bradykinin B1 receptor antagonist. Journal of Neuropathology and Experimental Neurology.
2005;
64
(9)
:
782-9
.
View Article PubMed Google Scholar -
Gabra
B.H.,
Sirois
P.,
Beneficial effect of chronic treatment with the selective bradykinin B1 receptor antagonists, R-715 and R-954, in attenuating streptozotocin-diabetic thermal hyperalgesia in mice. Peptides.
2003;
24
(8)
:
1131-9
.
View Article PubMed Google Scholar -
Zlotnik
A.,
Yoshie
O.,
Chemokines: a new classification system and their role in immunity. Immunity.
2000;
12
(2)
:
121-7
.
View Article PubMed Google Scholar -
White
F.A.,
Jung
H.,
Miller
R.J.,
Chemokines and the pathophysiology of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America.
2007;
104
(51)
:
20151-8
.
View Article PubMed Google Scholar -
Bhangoo
S.K.,
Ren
D.,
Miller
R.J.,
Chan
D.M.,
Ripsch
M.S.,
Weiss
C.,
CXCR4 chemokine receptor signaling mediates pain hypersensitivity in association with antiretroviral toxic neuropathy. Brain, Behavior, and Immunity.
2007;
21
(5)
:
581-91
.
View Article PubMed Google Scholar -
Wilson
N.M.,
Jung
H.,
Ripsch
M.S.,
Miller
R.J.,
White
F.A.,
CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain, Behavior, and Immunity.
2011;
25
(3)
:
565-73
.
View Article PubMed Google Scholar -
Menichella
D.M.,
Abdelhak
B.,
Ren
D.,
Shum
A.,
Frietag
C.,
Miller
R.J.,
CXCR4 chemokine receptor signaling mediates pain in diabetic neuropathy. Molecular Pain.
2014;
10
:
42
.
View Article PubMed Google Scholar -
Calcutt
N.A.,
Smith
D.R.,
Frizzi
K.,
Sabbir
M.G.,
Chowdhury
S.K.,
Mixcoatl-Zecuatl
T.,
Selective antagonism of muscarinic receptors is neuroprotective in peripheral neuropathy. The Journal of Clinical Investigation.
2017;
127
(2)
:
608-22
.
View Article PubMed Google Scholar -
Jolivalt
C.G.,
Frizzi
K.E.,
Han
M.M.,
Mota
A.J.,
Guernsey
L.S.,
Kotra
L.P.,
Topical delivery of muscarinic receptor antagonists prevents and reverses peripheral neuropathy in female diabetic mice. The Journal of Pharmacology and Experimental Therapeutics.
2020;
374
(1)
:
44-51
.
View Article PubMed Google Scholar -
Jolivalt
C.G.,
Han
M.M.,
Nguyen
A.,
Desmond
F.,
Alves Jesus
C.H.,
Vasconselos
D.C.,
Using corneal confocal microscopy to identify therapeutic agents for diabetic neuropathy. Journal of Clinical Medicine.
2022;
11
(9)
:
2307
.
View Article PubMed Google Scholar -
Sabbir
M.G.,
Calcutt
N.A.,
Fernyhough
P.,
Muscarinic acetylcholine type 1 receptor activity constrains neurite outgrowth by inhibiting microtubule polymerization and mitochondrial trafficking in adult sensory neurons. Frontiers in Neuroscience.
2018;
12
:
402
.
View Article PubMed Google Scholar -
Sabbir
M.G.,
Fernyhough
P.,
Muscarinic receptor antagonists activate ERK-CREB signaling to augment neurite outgrowth of adult sensory neurons. Neuropharmacology.
2018;
143
:
268-81
.
View Article PubMed Google Scholar -
Staskin
D.R.,
Robinson
D.,
Oxybutynin chloride topical gel: a new formulation of an established antimuscarinic therapy for overactive bladder. Expert Opinion on Pharmacotherapy.
2009;
10
(18)
:
3103-11
.
View Article PubMed Google Scholar -
Casselini
C.M.,
Parson
H.K.,
Frizzi
K.E.,
Marquez
A.,
Smith
D.R.,
Guernsey
L.,
A muscarinic receptor antagonist reverses multiple indices of diabetic peripheral neuropathy: preclinical and clinical studies using oxybutynin. Acta Neuropathologica.
2024;
147
(1)
:
60
.
View Article PubMed Google Scholar
Comments
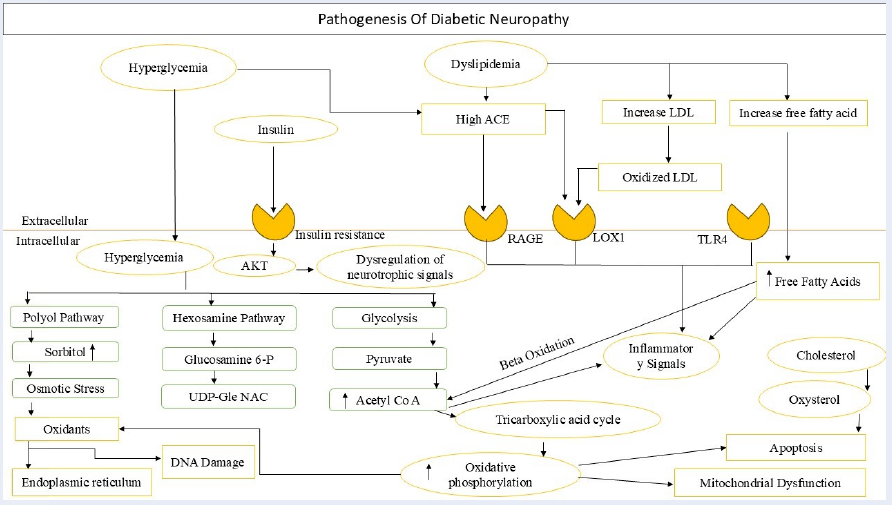
Article Details
Volume & Issue : Vol 11 No 8 (2024)
Page No.: 6698-6719
Published on: 2024-08-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 2798 times
- PDF downloaded - 988 times
- XML downloaded - 74 times
