

The Potential of Stem Cells in the Treatment of Diabetes: An Up-to-Date Review
- Department of Veterinary and Biomedical Sciences, College of Veterinary Medicine, University of Minnesota, Twin Cities, USA
- Department of Botany, Acharya Prafulla Chandra College, New Barrackpore, West Bengal, India
- Department of Microbiology, Swami Vivekananda Institute of Modern Sciences, Garia, West Bengal, India
- Department of Zoology, Krishnagar Government College, Krishnagar, West Bengal, India
- Medical Biotechnology lab, Faculty of Allied Health Sciences, Chettinad Hospital and Research Institute (CHRI), Chettinad Academy of Research and Education (CARE), Rajiv Gandhi Salai (OMR), Kelambakkam, Chennai, Tamil Nadu, India
- Department of Zoology, Acharya Prafulla Chandra College, New Barrackpore, West Bengal, India
Abstract
Introduction: Diabetes mellitus comprises a spectrum of metabolic disorders in which the body cannot adequately regulate blood glucose. In 2019, it ranked sixth among global causes of death, accounting for approximately 1.5 million fatalities. Current therapies rely on exogenous insulin or replacement of insulin-producing β-cells through whole-pancreas or isolated islet transplantation. Pluripotent stem cell (PSC)–based therapy offers a renewable source of patient-specific β-cells capable of restoring endogenous insulin production.
Areas Covered: Recent breakthroughs in directing PSCs—both embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs)—toward pancreatic lineages are summarized in this review. PSCs efficiently differentiate into functional, glucose-responsive β-cells as well as supportive islet cell types, broadening their therapeutic scope. Notably, patient-derived iPSCs created from diverse diabetic phenotypes can be gene-corrected and matured into insulin-secreting cells, paving the way for personalized medicine. Coupling PSC technology with CRISPR gene editing, 3-D organoid culture, and immune-evasive encapsulation devices is now moving first-in-human trials toward durable, insulin-independent outcomes.
Expert Opinion: Autologous PSC models not only enable mechanistic studies of diabetes pathogenesis but also guide precision drug discovery and cell-replacement strategies. To translate PSC therapy from bench to bedside, the field must still optimize differentiation yield, verify long-term safety, resolve immunogenic and ethical issues, and standardize manufacturing under Good Manufacturing Practice (GMP) conditions.
Introduction
Diabetes mellitus affected an estimated 537 million people worldwide in 2021 and, according to the International Diabetes Federation, this number could rise to 783 million by 20451. It imposes substantial premature morbidity and mortality. The disease is primarily divided into type 1 (T1D) and type 2 (T2D). T1D is an autoimmune disease in which autoreactive lymphocytes destroy pancreatic β-cells, eliminating endogenous insulin secretion2.
Individuals with T1D therefore depend on lifelong exogenous insulin. By contrast, T2D represents ~90 % of all diabetes cases and results from a combination of peripheral insulin resistance and insufficient compensatory insulin secretion by β-cells. Ongoing research continues to clarify the molecular mechanisms that drive T2D onset and progression3.
Monogenic forms of diabetes also exist, most notably neonatal diabetes mellitus (NDM) and maturity-onset diabetes of the young (MODY)4. NDM presents within the first six months of life and affects approximately 1 in 300 000–400 000 live births5, 6. MODY, an autosomal-dominant β-cell disorder that typically manifests in adolescence or early adulthood, accounts for <5 % of all diabetes cases7, 8. Molecular testing has so far delineated 14 MODY subtypes9.
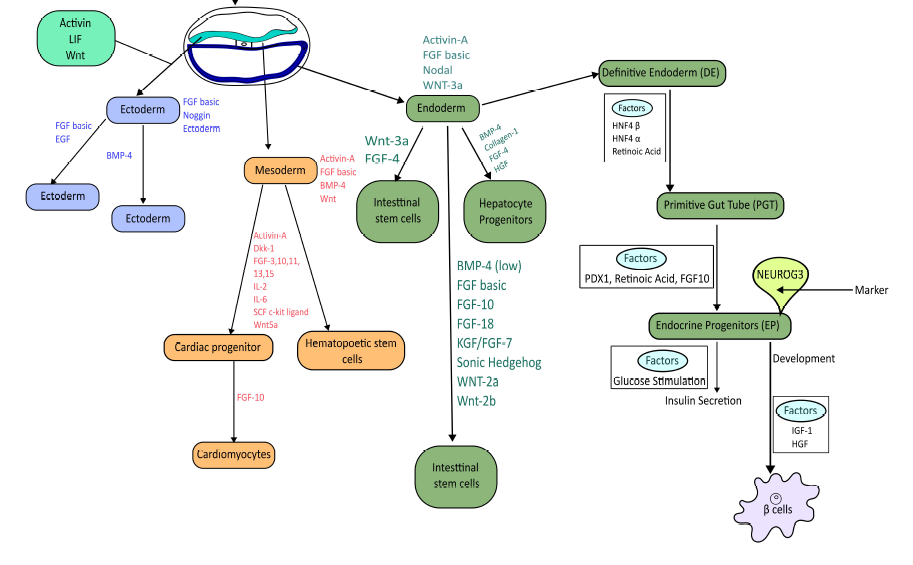
Because stem cells can modulate immune responses and differentiate into insulin-producing β-like cells, they are being explored as innovative therapies for diabetes10, 11, 12. Stem cells are classically categorized as totipotent, pluripotent, multipotent, oligopotent, or unipotent, depending on their developmental potential13. In a pioneering trial, Voltarelli et al. (2007) infused autologous hematopoietic stem cells (HSCs) into patients with recent-onset T1D and reported partial remission14. Subsequently, Bhansali et al. (2009) showed that bone-marrow-derived stem cells can safely improve β-cell function in T2D15. Numerous trials have since evaluated various stem-cell sources; however, the optimal cell type, dose, and delivery route remain to be defined, and severe infections have occasionally been observed16. Advances in stem-cell derivation and bioprocessing aim to provide an unlimited, donor-independent supply of transplantable β-cells for regenerative medicine17 (Figure 1).
Stem cell lineages have the potential to differentiate into β-cell lineages.
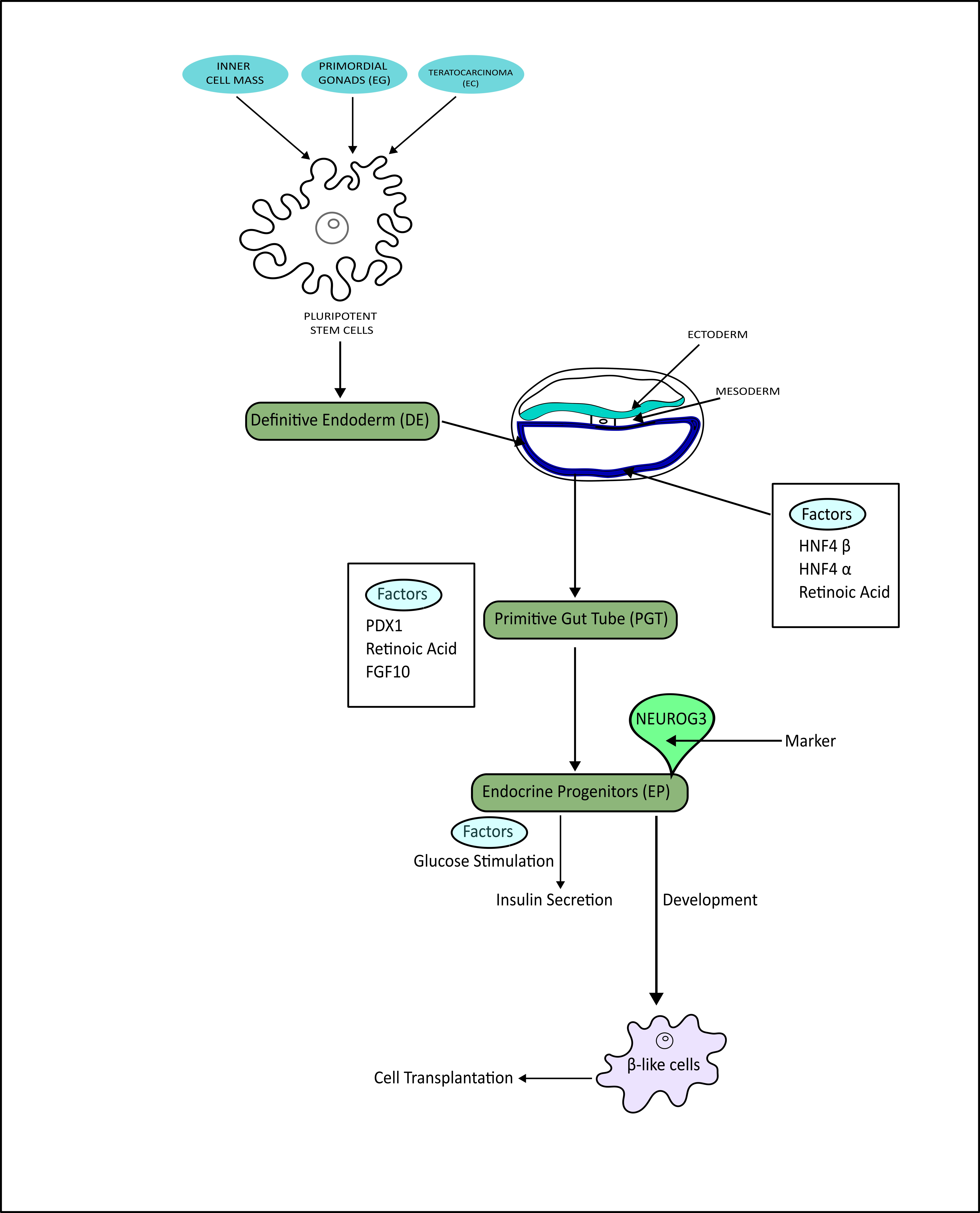
Embryonic stem cells (ESCs) are derived from the inner cell mass of the blastocyst, whereas induced pluripotent stem cells (iPSCs) are reprogrammed from somatic cells by ectopic expression of factors such as OCT3/4, SOX2, KLF4, and c-MYC. Both ESCs and iPSCs are pluripotent, meaning they can give rise to virtually any cell type, including cardiomyocytes, neurons, and pancreatic β-cells18, 19 (Figure 2).
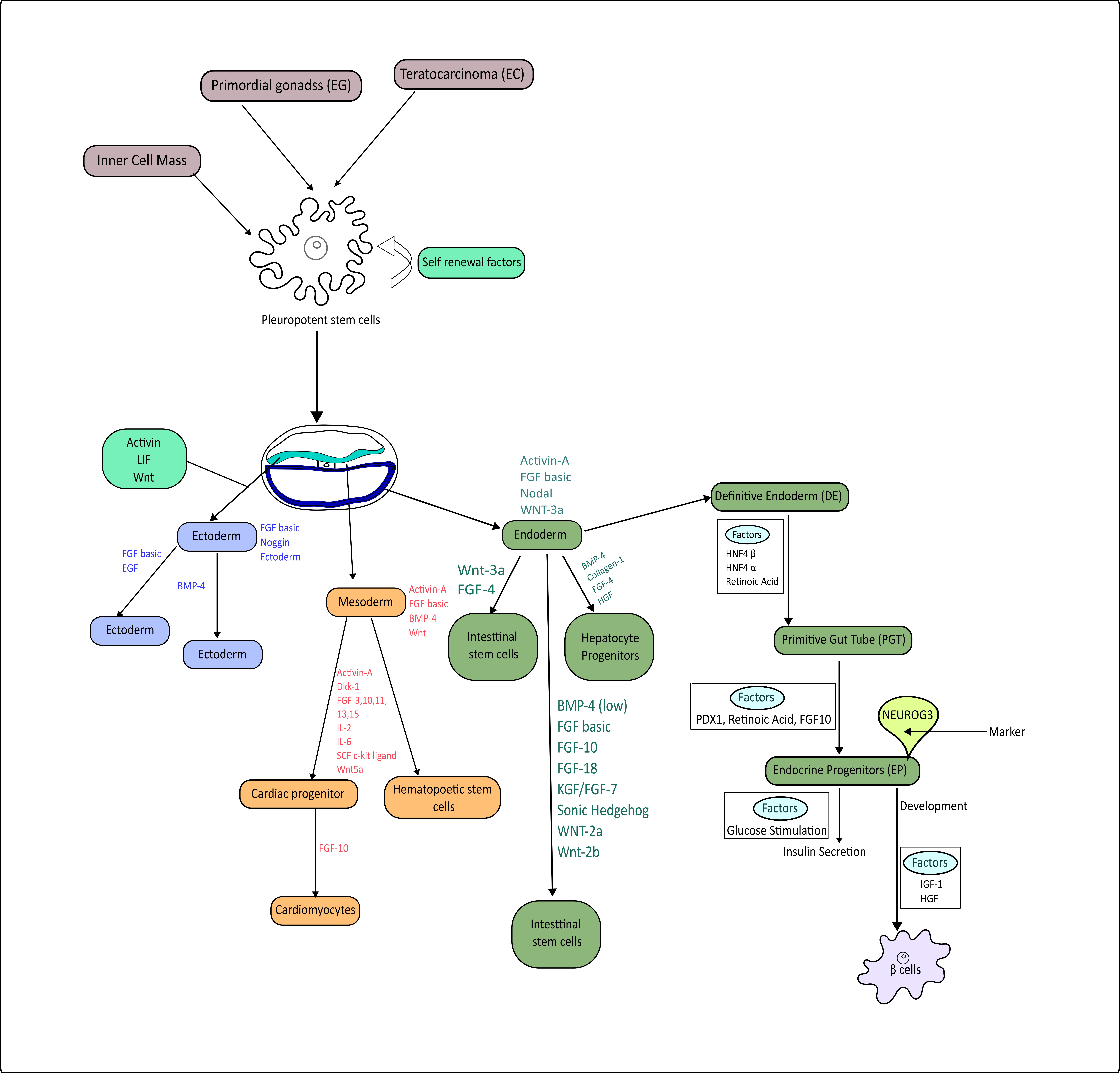
Markers specific to lineage and ESC-iPSC Differentiation Pathways.
Precision genome-editing, particularly CRISPR–Cas9, now allows targeted modification of transcription factors and signaling pathways in human pluripotent stem cells (hPSCs). Knockout of ARX, NKX6-1, or NEUROD1 clarifies endocrine lineage specification, whereas knock-in reporters such as PDX1-GFP or INS-mCherry permit real-time monitoring of β-cell maturation. Correction of pathogenic variants (e.g., HNF1A, INS) in patient-specific iPSCs paves the way for personalised cell-replacement therapies20. Single-cell RNA-seq and ATAC-seq provide high-resolution atlases of in-vitro differentiation, revealing rare progenitors, off-target populations, and regulators of β-cell maturation; only a fraction of derived cells fully resembles fetal or adult islets21. Engineering 3D microenvironments—vascularised islet organoids, hydrogel scaffolds, organ-on-a-chip systems—improves glucose responsiveness, endocrine maturity, and cell survival by recapitulating paracrine cues, extracellular-matrix signals, and biomechanical stiffness22. Together, these innovations are propelling stem-cell science from proof-of-concept studies toward robust, scalable, and clinically translatable regenerative therapies for diabetes.
Pathophysiology of Diabetes
Diabetes mellitus (DM) comprises a spectrum of metabolic disorders characterized by persistent hyperglycemia, yet the etiology and underlying mechanisms differ among subtypes. Appreciating these distinctions is critical to targeted prevention, accurate diagnosis, and effective therapy. The principal forms are type 1 diabetes (T1D), type 2 diabetes (T2D), gestational diabetes mellitus (GDM), and monogenic variants such as neonatal diabetes and maturity-onset diabetes of the young (MODY).
Type 1 Diabetes Mellitus (T1D)
T1D is a prototypic autoimmune disease in which autoreactive T lymphocytes progressively destroy pancreatic β-cells within the islets of Langerhans, culminating in absolute insulin deficiency. Both genetic susceptibility—particularly HLA-DR/DQ alleles—and environmental triggers (e.g., viral infections) initiate the immune assault. Pathological hallmarks include insulitis, β-cell apoptosis, and dense CD8⁺ cytotoxic T-cell infiltration23, 24, 25. Circulating islet-autoantibodies (against GAD65, IA-2, ZnT8, etc.) often precede clinical presentation and serve as predictive biomarkers. Oxidative stress and pro-inflammatory cytokine cascades further accelerate β-cell loss26. Because autoimmune activity usually persists after diagnosis despite intensive glycemic control, adjunctive immunomodulatory strategies are being explored.
Type 2 Diabetes Mellitus (T2D)
T2D arises from the synergy of peripheral insulin resistance and progressive β-cell dysfunction. Initially, skeletal muscle, liver, and adipose tissue become less responsive to insulin, prompting compensatory hyperinsulinemia. Hyperglycemia develops once β-cells can no longer sustain this output27, 28. Thus, T2D is defined by the dual defects of insulin resistance and relative insulin deficiency. Obesity-related inflammation, lipotoxicity, and mitochondrial dysfunction drive resistance; adipose-derived cytokines such as TNF-α and IL-6 impair insulin signalling, whereas chronic glucotoxicity induces β-cell exhaustion and epigenetic alterations29, 30.
Gestational Diabetes Mellitus (GDM)
GDM manifests when maternal β-cells cannot counterbalance the insulin-resistant milieu created by placental hormones (e.g., progesterone, human placental lactogen)31. Screening typically occurs at 24–28 weeks’ gestation32. Women with pre-existing β-cell impairment or risk factors—obesity, advanced maternal age, positive family history—are especially susceptible. Although glucose tolerance usually normalises postpartum, GDM unmasks an underlying metabolic vulnerability and substantially elevates future T2D risk33.
Monogenic Diabetes (MODY and Neonatal Diabetes)
Monogenic forms result from single-gene mutations that disrupt β-cell development, glucose sensing, insulin synthesis, or secretion, and are unrelated to autoimmunity or insulin resistance. In MODY, which is typically autosomal-dominant and presents in adolescence or early adulthood, hyperglycaemia is non-ketotic and non-insulin-resistant8. Common subtypes include GCK-MODY, characterised by mild, stable fasting hyperglycaemia due to impaired glucose sensing, and HNF1A-MODY, which shows progressive β-cell failure yet responds well to low-dose sulfonylureas34. Neonatal diabetes mellitus (NDM) appears within the first six months of life; mutations in KCNJ11 or ABCC8 blunt ATP-sensitive K⁺ channel closure, suppressing insulin release35. When such mutations are confirmed, high-dose sulfonylureas can replace insulin therapy36.
Secondary Diabetes
Secondary diabetes denotes hyperglycaemia caused by disorders or treatments that disturb glucose homeostasis rather than primary defects in insulin action or secretion. Examples include chronic pancreatitis37, haemochromatosis38, Cushing’s syndrome39, and prolonged exposure to glucocorticoids40 or atypical antipsychotics41. Mechanisms range from direct β-cell injury to severe insulin resistance at the receptor or post-receptor level.
The Vitality of β-Cells
Pancreatic β-cells within the islets of Langerhans orchestrate insulin secretion and are therefore central to glucose homeostasis. In diabetes, hyperlipidaemia, hyper-glycaemia and chronic inflammation converge to provoke endoplasmic-reticulum (ER) stress, oxidative stress and mitochondrial dysfunction, ultimately driving β-cell death and de-differentiation. Although ER and mitochondrial stress individually impair β-cell viability, recent work highlights their synergistic amplification of reactive oxygen-species (ROS) generation42, 43. β-cells inherently produce high ROS yet possess only modest antioxidant defences, rendering them exceptionally vulnerable to oxidative injury and functional collapse44, 45.
In type 1 diabetes (T1D) the immune system eliminates ≈90 % of β-cells, causing an early fall in insulin output that precedes overt hyper-glycaemia. Intriguingly, residual β-cells often persist in people with long-standing T1D46, implying that low-level endogenous insulin secretion can continue47, 48. In type 2 diabetes (T2D) approximately half of the original β-cell mass remains at diagnosis49, 50. Butler et al. analysed 124 human pancreata and found β-cell apoptosis was elevated ten-fold in lean T2D and three-fold in obese T2D, independent of auto-immunity51. These observations underscore the importance of preserving or restoring β-cell mass to maintain euglycaemia, making β-cell replacement a rational strategy for both T1D and T2D52. Whole-pancreas or islet transplantation can normalize glycaemia but is limited by donor scarcity, surgical risk and lifelong immuno-suppression. Consequently, stem-cell-derived β-cells have emerged as an attractive, potentially unlimited alternative source53, 54.
Key Features of Pluripotent Stem Cells (PSCs)
PSC populations—embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs)—combine unlimited self-renewal with the capacity to generate derivatives of all three germ layers. ESCs originate from the inner cell mass of the blastocyst, whereas iPSCs arise when somatic cells are reprogrammed by OCT4, SOX2, KLF4 and c-MYC expression (the Yamanaka factors)55. Pluripotency is sustained by interconnected transcription-factor networks, epigenetic regulators and signalling cascades including WNT, TGF-β/Activin/Nodal and FGF. This developmental plasticity underpins applications in regenerative medicine, disease modelling and high-throughput drug discovery. Nonetheless, genetic instability, tumourigenicity and line-to-line variability mandate rigorous quality control.
The 2006 discovery of iPSCs by Takahashi & Yamanaka revolutionised the field by providing an ethically acceptable ESC surrogate56, 57, 58, 59, 60, 61. Advances in vector design—from integrating retroviruses to non-integrating episomes, Sendai virus and mRNA—have enhanced reprogramming safety and efficiency62, 63, 64. Today, patient-specific iPSC lines enable precise disease modelling and personalised cell-based therapies, underscoring PSCs’ transformative potential65.
Directed Differentiation of PSCs
To generate functional cell types, researchers recapitulate embryogenesis in vitro using serum-free media, defined growth factors and stepwise signalling cues. For example, VEGF plus other angiogenic factors yield endothelial cells66; multistage Activin-A/retinoic-acid/Notch modulation directs PSCs to pancreatic β-like cells67; BMP4, FGF and HGF drive hepatic specification toward hepatocyte-like cells68; and 3-D culture of porcine iPSCs with retina-specific factors forms laminated retinal organoids69. Ongoing protocol optimisation is boosting purity, scalability and reproducibility, accelerating deployment of PSC-derived cells in drug screening, disease modelling and regenerative therapy.
Ethical and Clinical Issues in Pluripotent Stem Cell Technologies
Research involving pluripotent stem cells, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), holds tremendous promise for regenerative medicine and disease modeling; however, it is also accompanied by significant ethical and clinical challenges that demand careful analysis and innovative solutions70. The main ethical controversy surrounding ESCs arises from the need to destroy embryos during derivation, prompting intense debate over the moral status of the embryo and leading to diverse regional regulations that, in turn, shape funding, research priorities, and clinical translation71. Although iPSCs circumvent embryo destruction and have revolutionized the field, they introduce fresh ethical and safety concerns that likewise require rigorous oversight72.
Early‐generation iPSC protocols relied on oncogenic transcription factors, raising fears of tumorigenesis and underscoring the necessity for safer reprogramming methods that preserve genomic integrity73. Integration-free techniques now mitigate many of these risks, yet confirming both genomic and epigenetic stability in patient-derived iPSC lines remains a critical bottleneck before clinical deployment74. Because undifferentiated PSCs can form teratomas, transplantation therapies must meet stringent safety thresholds and demonstrate robust efficacy75. Even autologous iPSC-based therapies face variability in line-to-line quality, complicating standardization and quality control76, 77.
Transformation of Pluripotent Stem Cells into β-Cells that Secrete Insulin
Successful differentiation of PSCs into insulin-producing β-cells hinges on precise control of the culture microenvironment and a deep understanding of developmental signaling cues. Human ESCs, derived from the inner cell mass of blastocysts, possess unique epigenetic landscapes that preserve pluripotency; establishing stable hESC lines that faithfully recapitulate primary ESC characteristics is therefore essential for an unlimited cell supply78, 79.
Step-wise protocols have converted hESCs into insulin-secreting cells, initially achieving ~12 % efficiency but with limited glucose responsiveness. Subsequent optimization—such as fine-tuning in-vitro glucose levels and modulating growth factors like Transforming Growth Factor (TGF)—has boosted yields to ~25 %80, 81. These findings underscore the need for early-stage interventions that protect or regenerate endogenous β-cell mass, or for replacement strategies using engineered stem-cell-derived islet-like clusters82.
Standard differentiation proceeds through definitive endoderm (SOX17⁺), pancreatic progenitor (PDX1⁺), endocrine progenitor (NGN3⁺), and finally mature β-cell stages. At each step, specific factors are applied, and stage-specific markers confirm proper lineage progression83.
Stem cell transplantation and treatment
Transplantation studies indicate that the in-vitro microenvironment strongly affects pancreatic progenitor expansion and maturation. Using immature pancreatic progenitors rather than fully differentiated cells consistently enhances in-vivo β-cell development. For example, pancreatic precursors derived from hESCs become glucose-responsive, insulin-secreting β-cells after implantation under the kidney capsule or into adipose tissue of streptozotocin (STZ)-induced diabetic mice84. Likewise, iPSC-derived grafts placed in both T1D and T2D mouse models acquire glucose-regulated insulin secretion and reduce hyperglycaemia85. Transplantation of non-human-primate iPSCs achieves comparable glycaemic improvement in murine diabetes models86. In the NOD mouse, iPSC-derived insulin-producing cells transplanted into the kidney respond appropriately to rising glucose concentrations87. Collectively, these findings show that extensive pre-transplant in-vitro patterning is essential, whereas site-specific in-vivo cues complete endocrine maturation.
Differentiation of PSCs
β-cell differentiation from PSCs starts with induction of definitive endoderm (DE). Current protocols drive 60–80 % of hESCs toward DE that co-express SOX17, FOXA2, CXCR4, and GSC88. Activin-A–mediated Nodal signalling and canonical Wnt are the two dominant pathways. High-dose activin A (50–100 ng ml-1) in serum-free medium reliably generates DE and simultaneously exerts paracrine/autocrine survival effects on adult human islets89. Addition of sodium butyrate or PI3K inhibitors further boosts DE yield90.
Supplementing activin A with Wnt3A, CHIR99021 (a GSK3 inhibitor), or BMP4 can further improve DE induction, with CHIR99021 generally outperforming Wnt3A for SOX17/FOXA2 expression. Growth-and-differentiation-factor-8 (GDF-8, myostatin) as well as the small molecules IDE1/IDE2 can each convert ≈ 80 % of hESCs into DE cells91.
To steer DE away from hepatic fate and toward pancreatic lineage, BMP and FGF signals are usually blocked with Noggin and SU5402, respectively92. Cyclopamine (a HEDGEHOG inhibitor), FGF10, and Notch modulation (transient FGF10 followed by the γ-secretase inhibitor DAPT) are then sequentially applied to expand PDX1⁺ pancreatic progenitors and initiate endocrine commitment93.
Dorsomorphin (a BMP type-I receptor blocker) plus retinoic acid (RA) robustly induces PDX1⁺ progenitors, whose proliferation is sustained by epidermal growth factor (EGF). Indolactam V further enriches this population94, 95. Transition from PDX1⁺ to NGN3⁺ endocrine precursors is facilitated by SB431542 (a TGF-β receptor inhibitor) and VMAT2 inhibitors such as reserpine or tetrabenazine, ultimately yielding glucose-responsive β-like cells96.
Final maturation is promoted with forskolin, dexamethasone, hepatocyte growth factor, IGF-1, and GLP-1 analogues. Expression of NKX6.1 is mandatory: grafts with high NKX6.1 reverse hyperglycaemia in diabetic mice, whereas NKX6.1-low grafts do not97. Mature hESC/hiPSC-derived β-like cells co-express C-peptide, insulin, PDX1, MAFA, NKX6.1, NEUROD1, ISL1, and GLUT298, 99, 100.
Forced expression of key transcription factors can further enhance efficiency. PAX4 over-expression, for example, elevates INS, PDX1, GLUT2, and C-peptide transcripts101, whereas PDX1 or FOXA2 alone provide minimal additional benefit102, 103, 104. Despite these advances, fully mature, glucose-responsive β-cells remain difficult to obtain in vitro, and most protocols still yield cells with sub-optimal dynamic insulin secretion.
Differentiation of human-induced PSCs
hiPSCs follow a similar five-stage trajectory—SOX17⁺ DE → PDX1⁺ progenitor → NGN3⁺ endocrine precursor → NKX6.1⁺ immature β-cell → functionally mature β-cell105, 106, 107, 108. The first demonstration in 2008 used a four-step protocol to convert dermal fibroblast–derived hiPSCs into glucose-sensitive insulin-secreting cells109. Nevertheless, hiPSCs exhibit clone-to-clone heterogeneity: lines from T1D donors generate DE efficiently but diverge markedly at later pancreatic stages110, 111, 112.
Comparison of hESC- and hiPSC-based protocols
Both cell types rely on sequential modulation of Notch, BMP, Wnt, and TGF-β signalling and on core transcription factors PDX1, NGN3, and NKX6.1. However, hiPSCs often show variable NGN3 induction, greater signalling-pathway noise, and residual somatic epigenetic memory that can skew differentiation away from pancreatic fate113, 114, 115. Consequently, hESCs generally achieve higher C-peptide/insulin co-expression and superior glucose responsiveness, whereas hiPSC-derived β-like cells usually require extended in-vitro culture or in-vivo maturation to reach comparable function116, 117.
Succession Rate of β-cells
Efficient expansion of β-cell mass from endogenous sources requires simultaneously limiting β-cell apoptosis and stimulating new-cell formation. Finegood et al. (1995) quantitatively analysed β-cell turnover by BrdU/thymidine labelling in rat pancreas118. They calculated a daily turnover of ~2 % of β-cells in adult rodents. Extended BrdU exposure in adult mice showed that roughly 1 in 1,400 β-cells divides each day. Assuming no input from neogenesis, trans-differentiation or other sources, the daily growth rate equals 0.070 %119, 120. Even with zero β-cell death, replacing one-half of lost β-cell mass would therefore require ≈1,429 days—far longer than the average mouse life span. Human calculations are limited, as BrdU cannot be used ethically; Ki67 staining nevertheless indicates an even slower turnover that can rise several-fold during pregnancy121, 122.
An alternative strategy for diabetes therapy is to enhance endogenous β-cell renewal. Evidence shows that β-cell mass is plastic and adapts to changing secretory demand. Two main mechanisms are proposed: (i) replication of existing β-cells and (ii) differentiation of progenitors, possibly within the ductal epithelium. Replication has been documented in mice, rats and humans, and lineage-tracing in postnatal mice demonstrates that most new β-cells derive from pre-existing ones. The close anatomical relationship between β-cells and pancreatic ducts suggests a potential ductal progenitor source, but cross-sectional studies cannot yet pinpoint the exact origin of mature β-cells118, 119, 120 (Figure 3). Thus, insulin-positive cells observed near ducts in adult tissue may simply reflect residual patterns of fetal pancreas development rather than active duct-derived neogenesis119, 121, 122.
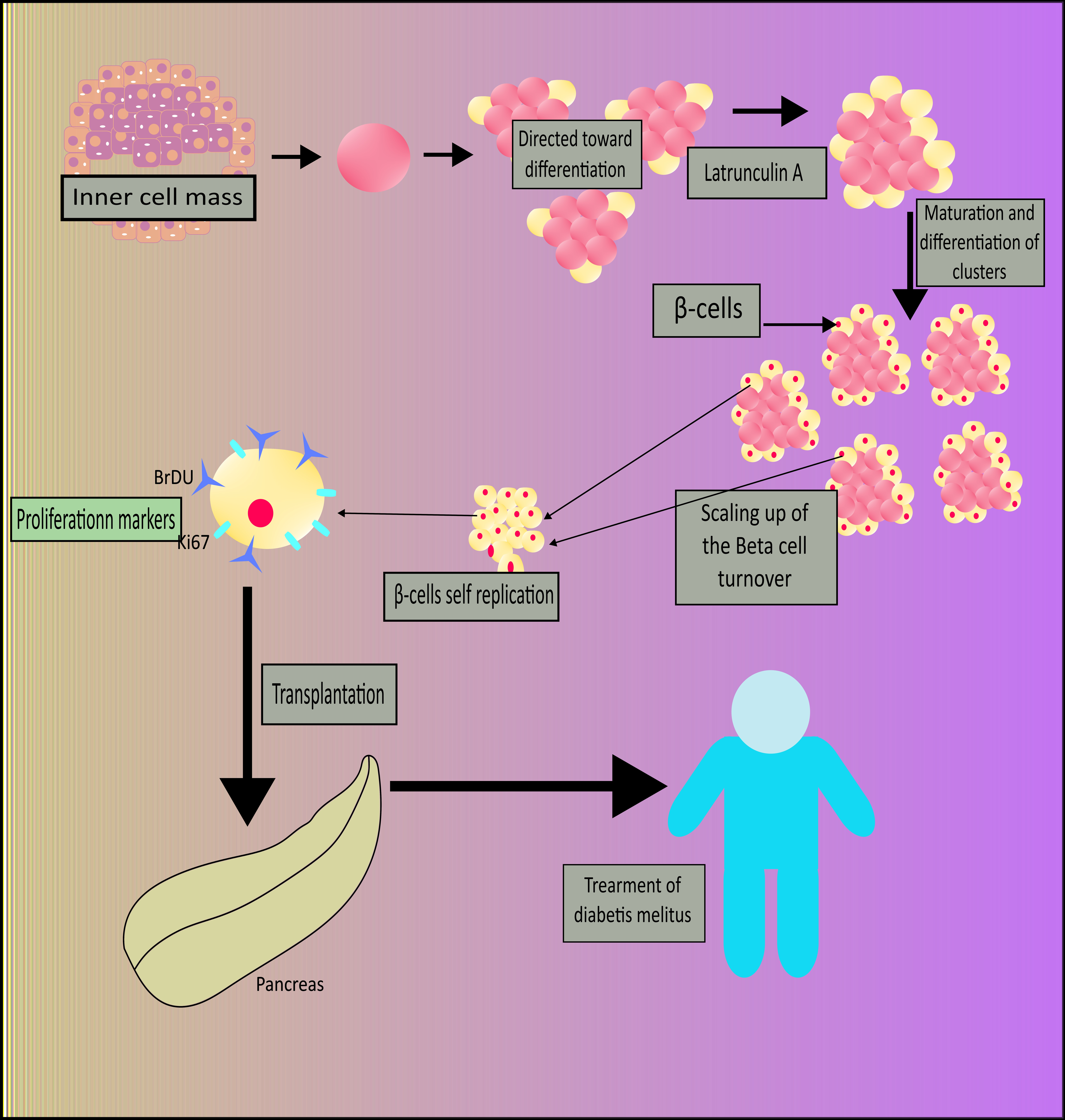
The regeneration of β-cells for treating diabetes mellitus.
Derivation of Patient-Specific Pluripotent Stem Cells for the Treatment of Diabetes
The pathogenesis of different diabetes subtypes is not fully understood. To address this gap, researchers generate patient-specific pluripotent stem cells (PSCs) from diabetic individuals as versatile in-vitro disease models. These cells can be differentiated into pancreatic lineages for mechanistic studies or transplantation, thereby providing new insights and enabling improved therapeutic strategies123.
Patient-Specific Embryonic Stem Cells
Somatic Cell Nuclear Transfer (SCNT), also known as therapeutic cloning, is used to create patient-specific embryonic stem cells (ESCs) from a patient’s somatic cells. In SCNT, a somatic-cell nucleus is transferred into an enucleated oocyte, producing an embryo that is nearly genetically identical to the donor123. Although SCNT first produced the cloned sheep Dolly in 1997, it is still not a routine method for generating patient-specific ESC lines. Recent breakthroughs have finally demonstrated successful reprogramming of human somatic cells into ESCs, after many unsuccessful attempts124. Notably, both hESCs and hiPSCs appear non-immunogenic after transplantation, supporting the concept of diabetes-specific ESC therapies123, 124. Nevertheless, SCNT is limited by ethical concerns and the scarcity of human oocytes. An alternative source of hESCs is embryos classified as abnormal during pre-implantation genetic diagnosis (PGD)123, 124, 125. PGD-derived hESCs have been exploited to model monogenic diseases in-vitro, but they cannot yield patient-specific ESCs for polygenic or idiopathic diabetes. Consequently, ethical restrictions and technical hurdles continue to hamper the broad use of SCNT and PGD for diabetes research123, 124, 125, 126.
Methods for Generation of Patient-Specific Pluripotent Stem Cells
Because hESCs face ethical, immunological, and logistical constraints, investigators increasingly focus on induced pluripotent stem-cell (iPSC) technology. Generating hiPSCs from diabetic patients and differentiating them into insulin-secreting cells provides a powerful platform for dissecting the earliest mechanisms of diabetes pathophysiology109, 127.
In the first study, hiPSCs were produced from skin fibroblasts of type-1-diabetes (T1D) patients using three transcription factors—OCT4, SOX2, and KLF4. More recently, hiPSCs were derived from participants with maturity-onset diabetes of the young (MODY) to model the disease in vitro. After identifying a heterozygous glucokinase (GCK) mutation, investigators generated MODY2-specific hiPSCs; their efficiency to form insulin-secreting cells was comparable to control lines because the mutation is hypomorphic127. In contrast, iPSCs harboring biallelic GCK inactivation showed markedly reduced β-cell differentiation128.
MODY2 patients possess β-cells with diminished glucose sensitivity. Correcting the GCK mutation in MODY2-hiPSCs restores normal β-cell glucose responsiveness, highlighting the utility of genome editing. Using a polycistronic lentiviral vector, other groups have derived hiPSCs from additional MODY subtypes without karyotypic abnormalities, facilitating the study of gene-specific contributions to pancreatic development and diabetes128.
Inter- and intra-patient variability in reprogramming efficiency and differentiation potential has been documented. For example, iPSCs from non-obese diabetic mice exhibited a compromised pluripotent state, underscoring the influence of genetic background16. Therefore, analysing multiple patient-derived lines alongside clinical data is essential to pinpoint disease-predisposing factors.
Traditional iPSC generation relies on integrating viral vectors that permanently insert reprogramming transgenes, raising risks of insertional mutagenesis and tumorigenicity. Non-integrating strategies—adenoviral delivery, Cre/LoxP excision, PiggyBac transposition, episomal plasmids, Sendai virus, synthetic mRNA and direct protein transduction—have been developed to overcome these issues129. Notably, integration-free hiPSCs have been produced from T1D and type-2-diabetes (T2D) patients; the Sendai viral genome is spontaneously lost after 8–12 passages while pluripotency is maintained127.
Eliminating viral transgenes reduces genomic alterations and makes iPSCs safer for cell therapy and disease modelling. Because some reprogramming factors (e.g., MYC) are oncogenic, protocols omitting them have been devised; for instance, hiPSCs from T1D patients were generated using OCT4, SOX2 and KLF4 without MYC130.
Utilising PSC-Derived Cells in Diabetic Mouse Models: A Treatment Strategy
Pluripotent stem cells (PSCs) possess the remarkable ability to differentiate into almost any cell type, making them indispensable tools for diabetes research65. Accordingly, diabetic mouse models are essential for dissecting disease mechanisms and testing emerging therapies. Through controlled in-vitro differentiation, investigators can generate insulin-producing pancreatic β-cells, endothelial cells, and immunomodulatory mesenchymal stem cells (MSCs) from PSCs131. When these PSC-derived cells are transplanted into diabetic mice, their effects on tissue regeneration, glycaemic control, and immune modulation can be quantified with precision132.
Transplantation of PSC-derived pancreatic β-cells offers the possibility of restoring endogenous insulin production and alleviating the consequences of insulin deficiency. Outcomes are monitored by evaluating cell survival, engraftment within host pancreatic tissue, and the re-establishment of normoglycaemia133. Such analyses are critical, as loss of functional β-cells is central to diabetes pathophysiology134.
PSC-derived endothelial cells additionally target the vascular complications of diabetes, supporting vascular repair and restoring blood flow to damaged tissues and organs135. Likewise, PSC-derived MSCs exert potent anti-inflammatory and immunomodulatory effects that counter the inflammatory milieu associated with diabetes. Mouse models allow researchers to quantify their capacity to dampen immune dysregulation, protect pancreatic cells, and accelerate tissue repair136.
Collectively, PSC-based interventions in diabetic mouse models refine our understanding of the disease and accelerate the development of next-generation therapies for patients.
Latest Advances in the Use of Pluripotent Stem Cells (PSCs) to Treat Diabetes
With its ability to precisely fix disease-causing mutations and improve cell function, CRISPR-Cas9 technology has become a cornerstone in the genetic engineering of PSCs. To restore normal insulin expression in iPSC-derived β-cells, CRISPR has been used to correct mutations in genes such as HNF1A and GCK, thereby restoring normal insulin expression in monogenic diabetes forms like MODY (Maturity-Onset Diabetes of the Young)137. Additionally, CRISPR is being investigated to generate universal, immune-evasive β-cell grafts for allogeneic transplantation by deleting immune-recognition molecules such as HLA.
PSC-derived three-dimensional (3D) pancreatic organoids have revolutionized both in vitro modeling and in vivo transplantation. These organoids mimic native islet architecture and function, including vasculature formation, cell-cell communication, and glucose-stimulated insulin release. Stepwise development of PSCs into mature β-cells within organoids is now possible thanks to protocols guiding differentiation through definitive endoderm, pancreatic progenitors, and endocrine precursors. To further enhance vascular integration and insulin-release kinetics, these organoids can be co-cultured with endothelial cells or embedded in extracellular-matrix hydrogels138.
The field is now entering the clinic, with multiple stem-cell-derived products in human trials. ESC-derived pancreatic islet cells are used in Vertex Pharmaceuticals’ VX-880 program, which has successfully restored insulin production in type 1 diabetes patients with undetectable C-peptide levels139. Similarly, ViaCyte’s PEC-Direct and PEC-Encap systems administer PSC-derived β-cell progenitors in encapsulated devices, aiming to achieve long-term insulin independence in early-phase trials and preclinical models140. However, foreign-body responses impaired graft vascularization and function in PEC-Encap (VC-01). In Phase 1/2 trials, PEC-Direct—an open device that permits vascularization but requires systemic immunosuppression—generated partial C-peptide and insulin secretion yet did not fully restore glycaemic control.
Patient-specific iPSC-derived β-cells are now being integrated into high-throughput drug-screening platforms to identify compounds that enhance insulin secretion or promote β-cell survival, creating a rapid feedback loop to clinical observation. The convergence of PSCs, CRISPR editing, and organoid technology is enabling high-throughput modeling of both monogenic and complex polygenic diabetes. Researchers now generate patient-specific, CRISPR-edited islet organoids to dissect gene function, predict therapeutic response and validate candidate drugs. These models recapitulate immune infiltration, ER stress, and β-cell dysfunction—hallmarks of both type 1 and type 2 diabetes.
Addressing Cell-Production Guidelines for PSC- and iPSC-Based Therapies
Strict adherence to Good Manufacturing Practice (GMP) is the cornerstone for translating pluripotent stem cell (PSC) and induced pluripotent stem cell (iPSC) products from the laboratory to the clinic. By controlling cell sourcing, scale-up, contamination risk, and batch-to-batch consistency, GMP safeguards product safety, reproducibility, and regulatory acceptance.
Recent literature therefore centres on the transition from bench-scale PSC cultures to pilot- and full-scale manufacturing. Huang et al. (2020) present a comprehensive GMP-compatible suspension-bioreactor workflow, detailing media optimisation, shear-stress management, and process automation required for clinical-grade expansion141. Similarly, Martins and Ribeiro (2025) describe the creation of GMP Master and Working Cell Banks, highlighting donor eligibility, traceability, and high-throughput quality control under EU and U.S. regulations142.
Thon and Karlsson (2017) show that feeder-free, xeno-free, closed-system bioreactors markedly improve reproducibility of platelet-producing PSC derivatives and facilitate regulatory approval143. Nath et al. (2020) further refine stirred-tank designs with in-line sensors, automated batch records, and continuous environmental monitoring, fully embedding GMP in iPSC expansion and differentiation144.
To minimise lot variability, Wong et al. (2017) introduced the CryoPause® workflow, which cryopreserves fully characterised PSCs in a ready-to-use format, enabling immediate and parallel differentiation across GMP sites145.
Demonstrating clinical relevance, Surendran et al. (2025) report a scalable, allogeneic retinal-pigment-epithelium (RPE) manufacturing process that meets FDA Investigational New Drug criteria via GMP-aligned cryopreservation, sterility, and endotoxin removal146. Likewise, Couture et al. (2014) provide detailed standard operating procedures (SOPs) for spinner-flask suspension culture that reliably generate undifferentiated PSCs at pilot scale147.
Difficulties and Challenges in Stem Cell Therapy for Diabetes
Pluripotent stem cells (PSCs) now allow researchers to reproduce diabetic pathology in vitro and to design cell-replacement strategies that were unthinkable a decade ago. Recent work has shown that patient-specific PSC lines can be differentiated into pancreatic lineages, providing platforms both for mechanistic studies and for the development of autologous therapies148, 149. Before these advances can reach the clinic, however, several obstacles must be overcome. First, robust, tumor-safe differentiation protocols are required to minimize the risk of teratoma formation149. Human embryonic stem cells (hESCs) additionally raise ethical concerns and problems of immune incompatibility, which currently restrict their clinical use. Somatic-cell nuclear transfer (SCNT) has recently been used to create hESC lines that are human-leukocyte-antigen (HLA) matched to individual patients, potentially reducing rejection150.
Although induced PSCs (iPSCs) represent a landmark step toward autologous β-cell replacement, important challenges remain. Comprehensive in-vitro assays and long-term transplantation studies are still needed to confirm the function, safety and durability of iPSC-derived β-cells. Genomic integrity is a key issue because some reprogramming methods employ integrating viral vectors that can introduce oncogenic or disruptive mutations150, 151. Even non-viral techniques can generate copy-number variations or point mutations that confound disease modelling; therefore, integration-free reprogramming and rigorous genomic screening should be standard practice.
Another hurdle is incomplete maturation. Cells produced in most differentiation protocols still express early developmental markers such as DPPA4, LIN28A and LIN28B, indicating a stage equivalent to < 6.5-week human embryos and explaining their poor glucose responsiveness152, 153. Refinement of stage-specific cues and 3-D culture systems is required to obtain fully mature, glucose-sensitive β-cells. Patient-to-patient and clone-to-clone variability further complicate individualized therapies; future work must elucidate and minimise these clonal differences.
Emerging evidence also overturns the assumption that autologous iPSC derivatives are intrinsically immune-privileged. Deuse et al. (2019) demonstrated that de-novo mitochondrial DNA (mtDNA) mutations can provoke T-cell-mediated rejection in syngeneic hosts154; similar observations were made by Sercel et al. (2021) and Bogomazova et al. (2024)155, 156. Hence, routine immunogenomic screening—especially of mtDNA—should precede any iPSC-based transplantation, even in autologous settings.
Whether iPSCs can fully replace hESCs remains unresolved, and the limited comparative data published so far underline the value of continuing hESC research alongside iPSC efforts127.
Conclusion and Future Prospects
PSC research continues to attract intense interest because these cells can generate isogenic models of diabetes and theoretically provide unlimited supplies of patient-matched β-cells. To translate this promise into therapy, investigators must eliminate undifferentiated contaminants, perfect maturation protocols, and address ethical and immunological barriers. The creation of SCNT-derived, HLA-matched hESCs and the advent of footprint-free iPSC technologies offer encouraging solutions. Combined with clinical-grade differentiation, 3-D islet-organoid platforms and precise CRISPR editing, PSC technology is progressing from glucose-control adjuncts toward durable, potentially curative interventions. Multiple allogeneic islet products are already in human trials, and genetically engineered, patient-specific β-cells are close behind. With continued multidisciplinary effort, PSC-based precision therapies are expected to transform diabetes management in the near future.
Abbreviations
3D: Three-Dimensional, ARX: Aristaless Related Homeobox, ATAC-seq: Assay for Transposase-Accessible Chromatin with sequencing, BMP4: Bone Morphogenetic Protein 4, BrdU: Bromodeoxyuridine, Cas9: CRISPR-associated protein 9, CD8⁺: Cluster of Differentiation 8 Positive, CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats, CXCR4: C-X-C Motif Chemokine Receptor 4, DE: Definitive Endoderm, DM: Diabetes Mellitus, DPPA4: Developmental Pluripotency Associated 4, EGF: Epidermal Growth Factor, ER: Endoplasmic Reticulum, ESCs: Embryonic Stem Cells, FDA: Food and Drug Administration, FGF: Fibroblast Growth Factor, FOXA2: Forkhead Box A2, GAD65: Glutamic Acid Decarboxylase 65, GCK: Glucokinase, GDM: Gestational Diabetes Mellitus, GDF-8: Growth and Differentiation Factor-8, GFP: Green Fluorescent Protein, GMP: Good Manufacturing Practice, GSC: Goosecoid Homeobox, GSK3: Glycogen Synthase Kinase 3, HEDGEHOG: a signaling pathway, HGF: Hepatocyte Growth Factor, HLA: Human Leukocyte Antigen, HNF1A: Hepatocyte Nuclear Factor 1 Alpha, hPSCs: Human Pluripotent Stem Cells, HSCs: Hematopoietic Stem Cells, IA-2: Insulinoma-Associated protein 2, IDE1/IDE2: Inducers of Definitive Endoderm 1 and 2, IGF-1: Insulin-like Growth Factor 1, IL-6: Interleukin-6, INS: Insulin, iPSCs: Induced Pluripotent Stem Cells, ISL1: ISL LIM Homeobox 1, KLF4: Krüppel-like factor 4, MAFA: MAF BZIP Transcription Factor A, MODY: Maturity-Onset Diabetes of the Young, MSCs: Mesenchymal Stem Cells, mtDNA: Mitochondrial DNA, MYC: Myelocytomatosis oncogene, NDM: Neonatal Diabetes Mellitus, NEUROD1: Neuronal Differentiation 1, NGN3: Neurogenin 3, NKX6-1: NK6 Homeobox 1, NOD: Non-Obese Diabetic, OCT3/4: Octamer-Binding Transcription Factor 3/4, PAX4: Paired Box 4, PDX1: Pancreatic And Duodenal Homeobox 1, PGD: Pre-implantation Genetic Diagnosis, PI3K: Phosphoinositide 3-Kinase, PSCs: Pluripotent Stem Cells, RA: Retinoic Acid, RNA-seq: RNA sequencing, ROS: Reactive Oxygen Species, RPE: Retinal Pigment Epithelium, SCNT: Somatic Cell Nuclear Transfer, SOPs: Standard Operating Procedures, SOX2: SRY-Box Transcription Factor 2, SOX17: SRY-Box Transcription Factor 17, STZ: Streptozotocin, T1D: Type 1 Diabetes, T2D: Type 2 Diabetes, TGF-β: Transforming Growth Factor Beta, TNF-α: Tumor Necrosis Factor Alpha, VEGF: Vascular Endothelial Growth Factor, VMAT2: Vesicular Monoamine Transporter 2, Wnt: Wingless-related integration site, ZnT8: Zinc Transporter 8
Acknowledgments
None.
Author’s contributions
All the authors have contributed substantially to the work. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have used generative AI and/or AI-assisted technologies in the writing process before submission, but only to improve the language and readability of their paper.
Competing interests
The authors declare that they have no competing interests.