

Aromatase inhibitors and their influence on brain – quick review
-
Poznan University of Medical Sciences

Abstract
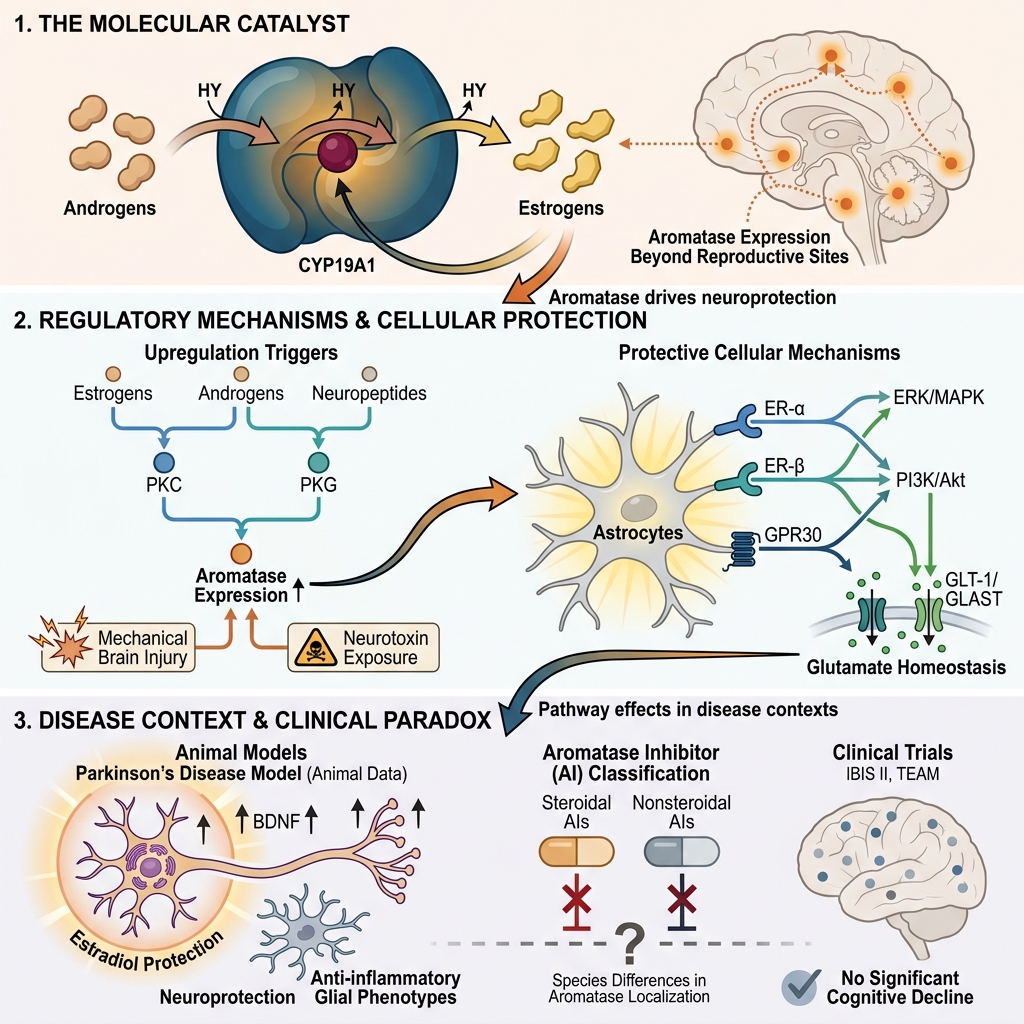
Aromatase, an enzyme responsible for converting androgens to estrogens, plays a crucial role in maintaining physiological homeostasis. It is also an essential factor in estrogen-dependent carcinogenesis. For many years, aromatase inhibitors (AIs), such as letrozole and vorozole, have been applied mainly in the treatment of breast cancer. Their utilization in managing breast and ovarian cancer, polycystic ovary syndrome, endometriosis, prostate diseases, and male infertility is well es-tablished. However, aromatase expression is not limited to classical estrogen-related tissues such as the breast, placenta, or ovaries. Its enzymatic activity is also observed in the brain, bones, and lungs. Consequently, this review focuses on the characterization of aromatase inhibitors, with particular emphasis on their adverse effects and the description of aromatase expression within the brain. The neuroprotective role of this enzyme, exhibited in response to mechanical damage and neurotoxins, is described with particular emphasis on the underlying physiological mechanisms. Furthermore, the clinical application of AIs and the role of aromatase in Parkinson's disease are also discussed.