
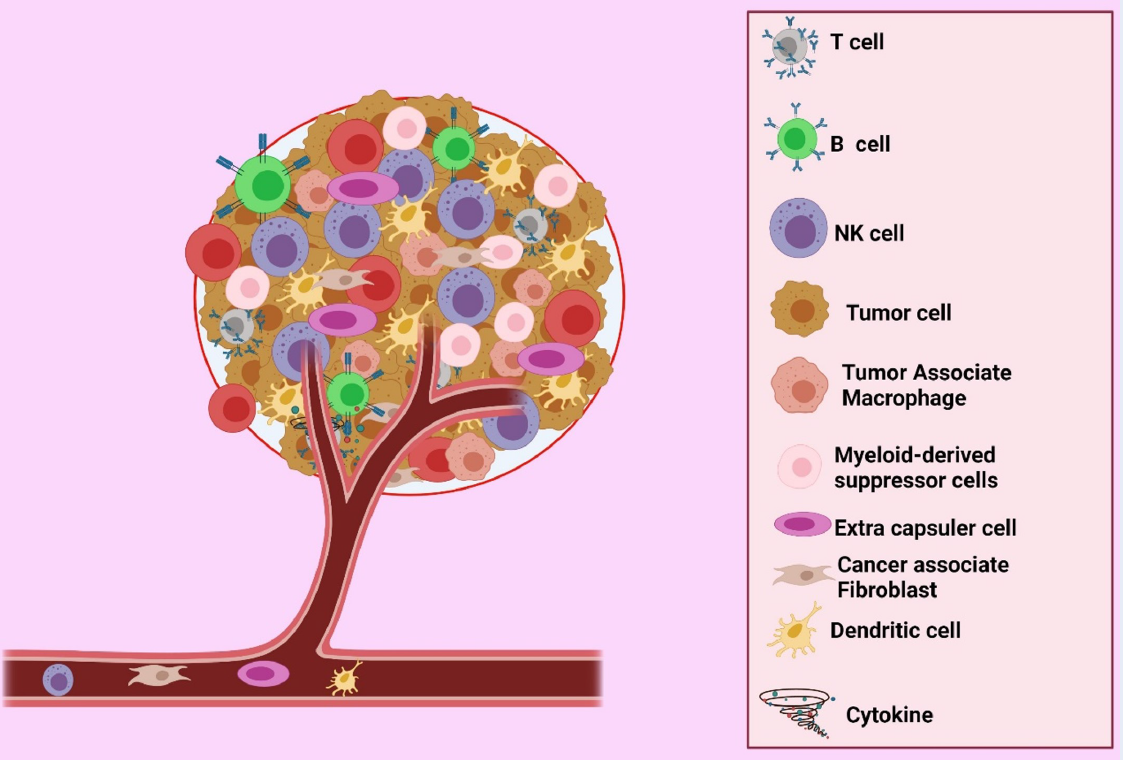

Overview of the advantages and disadvantages of chimeric antigen receptor T cell therapy in the tumor microenvironment
- Department of Zoology, University of Okara, Renala Khurd,56300, Pakistan
- Applied Molecular Biology and Biomedicine Lab, Department of Zoology, University of Narowal, Narowal, Pakistan
- Cell and Molecular Biology Lab, Institute of Zoology, University of the Punjab, Lahore-Pakistan
Abstract
T cells genetically modified to express a receptor that identifies a specific antigen, known as chimeric antigen receptor (CAR) T cells, have led to advancements in the treatment of hematological malignancies and the tumor microenvironment. CAR-T cells target the origin of tumors on the vascular side of inflammatory cytokines where the tumor microenvironment forms and block immunosuppressive checkpoints. The efficacy of CAR-T cell treatment remains controversial because its toxicity damages organ structures, including neurological, pulmonary, cardiac, and muscular structures, and causes fatal abnormality. In this review, we discuss the advantages and disadvantages of CAR-T cell immunotherapy in the tumor microenvironment.
Introduction
Cancer is the most crucial global economic and social burden. In 2012, more than 13.5 million new cancer cases were recorded, and more than 8 million affected patients died1. Malignant tumors are aggregates of cells that disrupt and impair the function of other normal cells in the body; progression from benignity to malignancy can impact health and cause death2. Tumors are a group of cancer cells whose microenvironmental makeup varies among types, but whose components generally include immune cells, blood vessels, and extracellular matrices. The tumor microenvironment (TME) is regarded as an active driver of cancer growth3. It is closely linked with cancer genesis, cancer cell propagation, survivability, tumor growth, and metastasis. The physiology involves blood vessels, inflammatory cells, immune cells, extracellular matrices, and biologically active molecules that interact with each other to form a tumor environment (Figure 1)4. Chronic swelling is a key risk factor of the development of tumors and is the major influencing factor of the formation of the TME that leads to metastasis5. The TME is directly linked with tumor development, growth, and metastasis and has an extensive impact on tumor treatment6.
The TME comprises various cellular components, including endothelial cells (ECs), that play a vital protagonistic role in tumor progression and fortification. The chief components of immune cells are granulocytes, lymphocytes, and macrophages, which are intricate in various responses, including inflammatory reaction and cancer development and progression. Tumor-associated macrophages can increase, arbitrate, or antagonize the anti-tumor activity of irradiation, cytotoxic mediators, and checkpoint inhibitors. Tumor cells can rove from the preliminary tumor site into the bloodstream and extend throughout the body via fibroblasts. Additionally, fibroblasts serve as a reliable conduit for ECs in tumors undertaking angiogenesis1.
Many anti-cancer medicines have been created to target critical signal molecules over-activated in malignant tissue. After the growing disclosure of an underlying immunosuppressive mechanism, immunotherapy is currently being evaluated as a new treatment strategy. However, attempts at curing cancer are frequently futile, as drug resistance appears inevitable. The concept of a TME can be viewed as an inexplicable means of explaining why many cancer treatments eventually fail. Accordingly, the fundamental method is known to involve de novo mechanisms, in which tumors either offer a new immortal signal for a cell or adjust specific default signaling pathways, thereby avoiding the influence of original drugs and causing resistance5.
Some drugs have been previously used to target the TME, and many of them currently remain under observation or processing in pre-clinical trials. At the end of the 20 century, cisplatin was used as an anticancer therapy; thereafter, its application was withdrawn owing to its high ototoxicity and emetogenic adverse events7. A variety of cellular therapies for cancer have been introduced, including chimeric antigen receptor (CAR) T cell treatment targeting solid TMEs. In this review, the clinical use and different experimental approaches of CAR-T cell treatment are described along with their background.
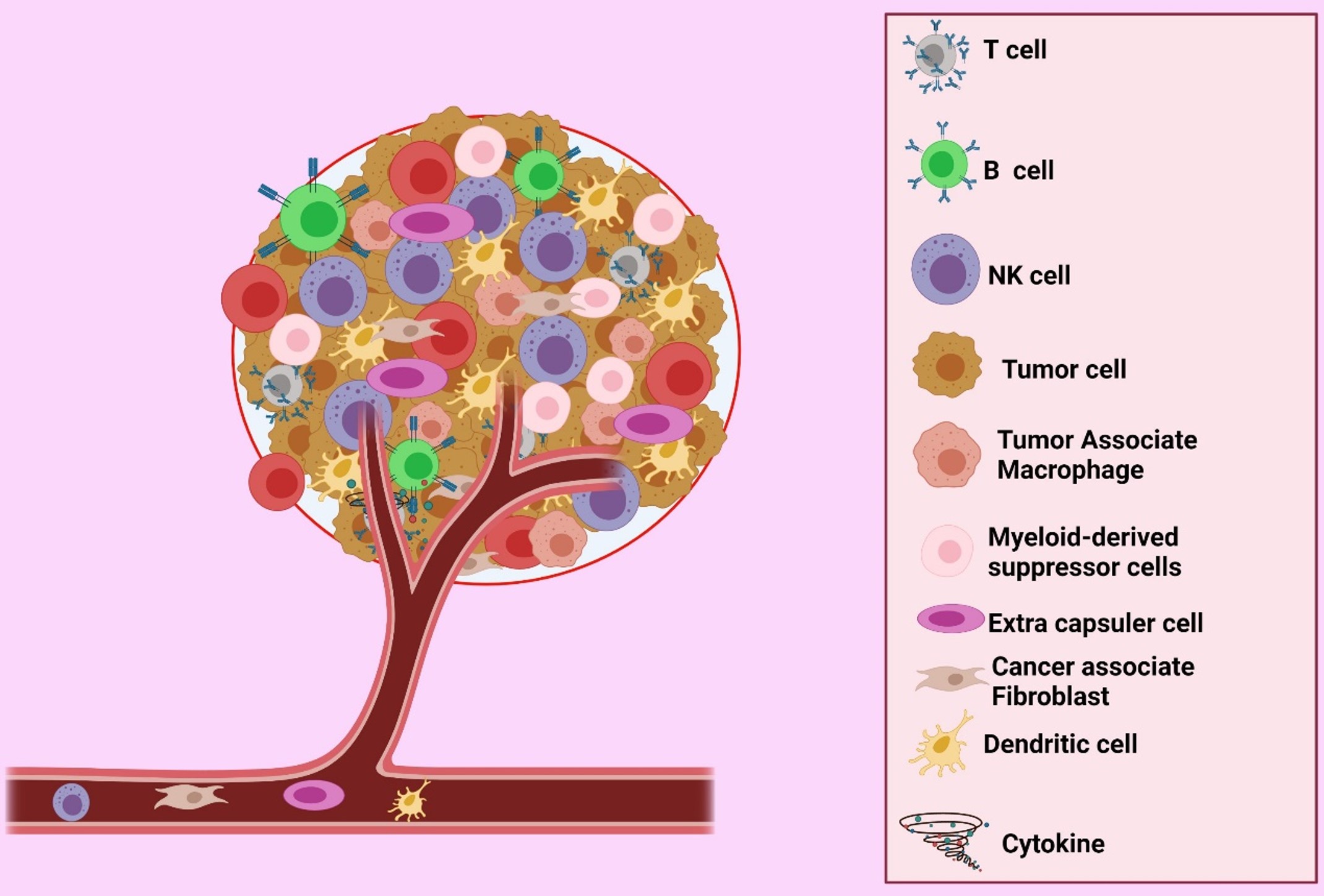
Intracellular and extracellular components of Tumor microenvironment. Tumor Microenvironment (TME) includes the T cells, B cells, Natural killer (NK) cells, Tumor cells, Tumor-Associated Macrophages (TAM), Myeloid-derived suppressor cells (MDSCs), Extra capsular cell (EC), Cancer associate Fibroblast (CAF), Dendrite Cell (DC), and Cytokines.
CARs
CARs were the first set of antigen receptors extensively researched for use in T cell genome modification. They consist of a particular binding domain that detects and binds a specific intracellular domain antigen transferring cellular signals. Cancer cells may be targeted through CARs for cancer immunotherapy. In continuing clinical investigations, four generations of CARs have been discovered on the basis of the intracellular signaling region. The first type of CARs includes only CD3ζ as an internal cellular signaling transduction area. In contrast to second-generation CD3ζ CARs, co-stimulating domains include CD28, 4-1BB (CD137), CD27, or OX40. In the third generation, CD3ζ assembly and two subdomains—CD28 and 4-1BB—are involved and decrease the constitutional symptoms (high fever, malaise, fatigue, and myalgia) of cytokine release syndrome (CRS)8. Zhao et al. described that CARs with mutual CD28 and 4-1BB co-stimulatory colonies increased CD4 and CD8 T cell augmentation and amended B cell acute lymphoblastic leukemia and tumor regression in xenograft models9.
CARs might be altered through the inclusion of additional genes, such as those encoding the strong cytokines of anti-tumor therapies (i.e., IL-2, IL-5, and IL-12), which can lead to the enlargement of “armor-powered” fourth-generation CAR-T cells10. The fourth type of CARs is known as the T cells redirected for universal cytokine-mediated killing (TRUCK); it is formed in relation to second-generation constructs by adding IL-1211. TRUCK boosts T cell activation while also activating and attracting distinctive immune cells to destroy antigen-ve in the targeted region. It would be interesting to discover the protagonistic role of TRUCK in influencing the tumor atmosphere over the transcriptional discharge of recombinant immune transformers11, 12. Many kinds of viral pathogenic infections, metabolic illnesses, and autoimmune disorders can be treated with TRUCK11. These succeeding generations of CAR-T cells have gained much interest in tumor treatment13.

The complete journey of development in the treatment of leukemia, lymphoma, and Approval of CAR-T by the FDA and the continuing journey of immunotherapy.
Role of CAR-T cells in the TME
T cells genetically engineered to prompt CARs are a promising new cancer treatment14, 15. This strategy was a breakthrough in the field of oncology in mid-2017. The FDA has permitted the use of CAR-T cell adoptive therapy in pediatric and adult patients with lymphoblastic leukemia with a complete response rate of 75% to 90% (Figure 2). After one and a half months, CAR-T cell therapy was also recommended for B cell lymphoma and malignancies16. The process does not halt once a CAR-T cell finds its way inside the TME. The TME has been widely described as hostile against T cells. The glycolytic metabolism of tumor cells makes them harmful, acidic, low-nutrient, and oxidative to the environment17, 18. Some means to modify the TME to progress the efficiency of CAR-T cell treatment have been developed; many other cells may also assist in improving tumor intolerance to another type of immunotherapy. Several concepts addressed are related to and have been clarified by investigations of adoptive cell treatment, including tumor-infiltrating lymphocytes (TILs) and TCR-engineered T cells19.

Summary of antigens targeted on different tumors by CAR-T cells registered at clinicaltrials.gov.
Attacking hypoxia and metabolism
Low oxygen concentrations in the blood are known as hypoxia and are the key internal environmental stressor involved in tumor progression. The clinical hazard is tumor cell oxygen scarcity20. Hypoxia has been shown to activate genes implicated in the directive of cellular differentiation, extracellular matrix formation, cellular binding, and other tumorigenic features. The induction of the hypoxia-inducible factor (HIF) family of transcription factors is widely used to achieve these effects. HIF-1, 2, and 3 are members of this family that regulate vessel formation, cell proliferation, and tissue transformation as in physiological processes upon exposure to low-slung oxygen levels. Further, a set of prolyl-4-hydroxylases and hydroxylate HIF-1 are involved with two conserved residues—proline 402 and proline 564—under normal oxygen circumstances21, 22. Hence, hypoxia is tremendously associated with tumor metastasis because it increases the metastatic potential of tumor cells, escalates the genomic uncertainty, and stimulates anaerobic metabolism and angiogenesis23, 24, 25. It is also found in bone marrow hematopoietic niches containing B lineage cells26. Hypoxia has been shown to impede anti-tumor immune retort via many mechanisms. Upregulation of PD-L1 expression in tumor cells and MDSCs is a crucial approach27, 28. Tumor cells can enhance their proliferation by suppressing immune responses via upregulation of reactive oxygen species production in the TME. Accordingly, Ligtenberg reported inserting catalase (CAT) into CAR-T cells to increase their anti-oxidative ability through HOmetabolism and consequently progress the persistence of the anti-tumor effect of CAR-T cells. This CAT alteration lowered OS in the TME and improved CAR-T cell anti-tumor activity29. Curran and colleagues reprogrammed the TME to amplify the anticancer effect of CAR-T cells by assisting in the conscription of an endogenous immune response, which is an established manifestation of CD40L by CAR-T cells30.

Approaches for overcoming CAR-T cell obstacles in the solid tumor microenvironment.
Objective (immune checkpoints) of CAR-T cells
Immune checkpoints are a group of co-stimulatory and repressive receptors implicated in the T cell response mechanism. They are critically aimed at the modulation and balance of the intensity with the purity of the T cell response within homeostasis maintenance conditions, which contributes to the preservation of self-tolerance and avoidance of irrational response tissue impairment. Nonetheless, inhibitory immunological barriers, including lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), SHP-1, and CTLA-4 in T cells, could suppress the cell properties31.
Show the global trials of CAR T in a different area of the tumor microenvironment to evaluate the therapeutic effects of CAR-T cells (registered at clinicaltrials.gov/)
|
Sr. No |
Trial Numbers |
Type of tumors |
Antigen targets |
Phases |
Enrolled populations |
|---|---|---|---|---|---|
|
1 |
NCT05373147 |
Solid Tumor |
Mesothelin |
I |
21 |
|
2 |
NCT05089266 |
Colorectal cancer |
Mesothelin |
I |
30 |
|
3 |
NCT03179007 |
Advanced Solid Tumor |
Mucin-1 |
I/II |
40 |
|
4 |
NCT03182816 |
Advanced Solid Tumor |
EGFR +ve |
I/II |
40 |
|
5 |
NCT02930993 |
Mesothelin Tumors |
Mesothelin |
I |
20 |
|
6 |
NCT05248048 |
Metastatic Colorectal Cancer |
NKG2D |
I |
09 |
|
7 |
NCT03030001 |
Solid Tumor Advanced Cancer |
Mesothelin |
I/II |
40 |
|
8 |
NCT03747965 |
Solid Tumor |
Mesothelin |
I |
10 |
|
9 |
NCT04556669 |
Solid Tumor, Sarcoma, Cervical Cancer, NSCLC |
PD-L1 CD22 |
I |
30 |
|
10 |
NCT03932565 |
Nectin4-positive Advanced Malignant Solid Tumor |
Nectin 4-positive |
I |
30 |
|
11 |
NCT03182803 |
Advanced Solid Tumor |
CTLA-4/PD-1 |
I/II |
40 |
|
12 |
NCT04665076 |
Plasma Cell Tumors |
MSLN +ve |
I |
60 |
|
13 |
NCT04503980 |
Colorectal cancer Ovarian Cancer |
αPD1-MSLN |
I |
10 |
|
14 |
NCT05420545 |
Metastatic Tumor, Solid Tumor, Renal Cell Carcinoma, Ovarian Cancer, Cervix Cancer |
CD70 |
I |
36 |
|
15 |
NCT02547961 |
Breast Cancer |
HER-2 |
I/II |
0 |
|
16 |
NCT03545815 |
Solid Tumor |
PD-1 and TCR |
I |
10 |
|
17 |
NCT05123209 |
Liver Cancer |
IM83 |
I |
12 |
|
18 |
NCT04283006 |
Lymphoid Hematological Malignancies |
CD20 CD22 |
I |
100 |
|
19 |
NCT02915445 |
Malignant Neoplasm of Nasopharynx TNM Staging Distant Metastasis (M) |
EpCAM |
I |
30 |
|
35 |
NCT02958397 |
Myeloid Malignancies |
CD33 |
I,II |
45 |
|
21 |
NCT04513431 |
HER-2 Gene Amplification, Gastric Cancer Breast Cancer, Overexpression •Solid Tumor |
HER-2 |
I |
220 |
|
22 |
NCT03706326 |
Hematologic Malignancy |
MUC1 |
I/II |
20 |
|
23 |
NCT02442297 |
Advanced Esophageal Cancer |
HER-2 |
I |
28 |
|
24 |
NCT04790747 |
Hematological Malignancies |
CAR-T with Radiotherapy |
I/II |
50 |
|
25 |
NCT05211557 |
Ovarian Cancer |
B7H3 |
I/II |
15 |
|
26 |
NCT02958384 |
Myeloid Malignancies |
Lewis -Y |
I,II |
45 |
|
27 |
NCT03874897 |
Advanced Solid Tumor |
Claudin18.2 |
I |
123 |
|
28 |
NCT03330834 |
Advanced Lung Cancer |
PD-L1/PD-1 |
I |
1 |
|
NCT05420545 |
Metastatic Tumor, Solid Tumor, Renal Cell Carcinoma, Ovarian Cancer, Cervix Cancer |
CD70 |
I |
36 | |
|
30 |
NCT04287309 |
Hematological Malignancy |
CD19 |
NA |
20 |

The summary of toxicity related to body organs affected by CAR-T cells included Neurological toxicity, Cardiac abnormality, and Immunological compromising condition that also affects nephrotoxicity.
LAG-3
CD223 is recognized as LAG-3, an immunosuppressive agent extremely articulated in triggered CD4 and CD8 T cells and Tregs32. LAG-3 escalates the activity of CAR-T cells upon antigen arrangement of the mark and the TME after CAR-T cell therapy33. CAR-T cells substantially increase the expression of LAG-3 in CD4 and CD8 T cells. They instigate precise expression of PD1 and demonstrate adaptive activation of immune checkpoints simultaneously with striking elevations in the expression of LAG-334. The effectiveness of CAR-T cell therapy can also be boosted by the combination of LAG-3 and PD-1.
TIM-3
TIM-3 expresses similarly to PD-1 in T cells within the TME, which becomes a co-inhibitory binding site, suppressing T cell proliferation and cytokine emission35. The TIM-3 levels in cancer cell antigen-specific CD8 T lymphocytes and CD8 TILs are increased in patients with cancer. Anti-TIM-3 monoclonal antibodies can stimulate pathways in addition to T cell activation, leading to tumor antigen-specific T cell proliferation and cytokine production36 . An increased TIM-3 expression might be a pathway of adaptive tumor resistance to PD-1 restriction therapy, and multimodal inhibition of the PD-1 and TIM-3 pathways may be beneficial in reversing T cell deprivation and establishing anti-tumor immunity37. In mice with relapsed acute myeloid leukemia, there is considerable amplification of TIM-3 in CAR-T cells compared with T cells extracted during healing after CAR-T cell therapy. Moreover, combining systemic PD-1 or TIM-3 blockage with CAR-T cell therapy has been reported to yield significant anticancer activity, implying that TIM-3 blockage might be helpful in association with cell therapy36.
Targeting cytokines through CAR-T cells
Inducing the native discharge of stimulatory substances to boost anti-tumor immune function is another possible approach for modifying the TME to maximize adoptive cell therapy efficacy. IL-12 and IL-18 are two viable options. IL-12 is an inflammatory cytokine capable of increasing T cell excitation, inducing Th1 CD4 T cell responses and CD8 clonal expansion, and playing an effector role38, 39. In addition to emission with CAR production by fourth-generation CAR-T cells—the quintessential impressive cytokine for TRUCK, IL-12 promotes tumor regression via at least three mechanisms: CAR-T cell progression and preservation40, 41 and tumor eradication decreased by IL-12 secreting CAR-T cells42. Meanwhile, IL-18 reduces the toxicity level owing to lesser production of TNF-α and increases TILs by diminishing macrophage conscription, thus fabricating innocuous and higher anti-tumor activity levels43. IL-12 stimulates both Th1 and Th2 cells, and IL-18 boosts instinctive immune cell bustle when combined with IL-12, providing a rationale for inducible IL-18 TRUCK T cells44. In pre-clinical studies (NCT00924326, NCT00019136, NCT04119024, and NCT03098355), IL-2, the single γc cytokine licensed by the FDA, was intensively examined in relation to intravenous or subcutaneous CAR-T cell treatment for malignancies and was found to be capable of promoting the growth of adoptive immunological cells45.
IL-4, which collaborates with IL-10 and TGF-α and provokes macrophage induction, is another immunosuppressive cytokine. Chimeric receptors engineered to articulate the IL-4 receptor ectodomain provide active signals that mimic IL-2 or IL-7 receptors and convert the suppressive function of IL-4 into a stimulatory function46, 47. An inverted cytokine receptor has been used to treat prostate cancer based on increased IL-4 intensities (ICR). The extracellular area of the inhibitory IL-4 receptor is coupled with the intracellular immunostimulatory domain of IL-7 to produce this 4/7 ICR. The co-expression of an anti-PSCA first-generation CAR vector and an ICR improved the anticancer activity in vitro and in vivo. This method converts an inhibitory signal for T cells into a stimulatory signal while depriving cancer cells of a key growth factor. An amalgamation of the second type of CAR and 4/7 ICR might be tested to improve these results47.
Tumor vasculature
Anti-angiogenic therapy is currently widely used for cancer treatment. However, achieving longevity with angiostatic or vascular-targeted medicines remains challenging48. Thus, a superior approach would be to precisely object the vascular structure using tumor endothelial biomarkers. The development of planned T lymphocytes armed with CARs pre-defines the specificity of CAR-T cells49. The anti-inflammatory microenvironment inside the tumor must be overcome by T cells. Immune exploitive cytokines, including IL-10, TGF, and VEGF, as well as directing T cells and MDSCs, are abundant in the TME, enabling even destructively activated anti-tumor cytotoxic T cells to become dormant50. Prostate-specific membrane antigen, αvβ3 integrin, TEM8, EIIIB, a splice form of fibronectin, and CLEC14A101 have also been previously investigated. These markers are overexpressed in the vasculature of a wide range of compacted human malignancies51. Several methods define cancerous ECs lining the vessels that explicitly constrain tumor immunity, such as over-declustering of ICMA-1 and VCAM-1, which are vital for a series of steps separate from leukocyte adherence to ECs and consequent diapedesis. Effector cells are impotent to transverse the ECs hooked on the tumor bed52. T cell strategies for solid tumors entail selecting analyses that are upregulated in the tumor vasculature. Epithelial tissue-like vessels targeting the tumor vascular structure matrix boundary might be useful, since an impaired vascular integrity may increase collateral immunity51.
Bellone and his colleague reported that the combination of chemotherapy with aggressive immunotherapy targeting the vasculature is another beneficial strategy. Notably, frequent administration of smaller doses of conventional cytotoxic chemotherapeutics, such as cyclophosphamide, could induce stress on the endothelium and demonstrate anti-angiogenic functions, including over enhancement of thrombospondin-153.
Selecting target molecules with a higher abundance at the luminal portion of the tumor endothelium may boost arrangement and thus accessibility of CAR-T cells. Using CAR-T cells with CARs that recognize well over one objective can improve attraction and hence the distinctiveness and protection of modified T cells toward this vascular structure formation.
Toxicity of CAR-T cells
Neurological toxicities, including aphasia, tremor, ataxia, myoclonus, and CRS, are related to the usage of CAR-T cell therapy. When CAR-T cells cross-react with an antigen expressed in normal tissue identical to the target antigen expressed by cancer, organ harm could theoretically ensue in CARs. Clinical trials have not documented such toxicity, but which has been observed in other clinical trials of CARs54. In most cardiovascular cases, tachycardia also occurred in patients with high-degree fever55; in some other cases during trials, cardiac ejection and a contrary higher level of serum troponin were observed56.
Many other patients also report hypoxia, dyspnea, and pneumonitis57. CRS is a multi-organ complicated toxicity syndrome that includes nephrotoxicity, hepatotoxicity, musculoskeletal toxicity, and hematological and multi-organ consequences54. The toxicity of CAR-T cells is summarized in Figure 5.
Conclusion
Solid TMEs are complex and vary in characteristics, and a solitary therapy may provide a partial clinical effect. CAR-T cells can kill tumor cells directly. The combination of CAR-T cell therapy with other approaches to overcome various hurdles in the TME can potentially yield excellent tumor-killing effects while diminishing CAR-T cell-induced organ toxicity.
Abbreviations
CAF: Cancer associate, CAR-T: Chimeric antigen receptor T cell, CASC: Cancer-associated cells, DC: Dendritic cell, ECC: Extra capsular cell, ECs: Endothelial cells, HIF: Hypoxia-inducible factor, LAG-3: Lymphocyte activation gene 3, MDSCs: Myeloid-derived suppressor cells, NK: Natural killer (NK) cells, TAM: Tumor-Associated Macrophages, TGF: Transforming growth factor, TILs: Tumor- infiltrating lymphocytes, TME: Tumor microenvironmnet, TRUCK: T-cells redirected for universal — mediated killing
Acknowledgments
The authors thank full to the Vice-Chancellor University of Okara. All figures were originally drawn on Biorender.com.
Author’s contributions
All authors in this current article sufficiently contributed to the conceptualization, design of the manuscript by (Adil Farooq), editing by (Muhammad Babar Khawar), and revision by (Muddasir Hassan Abbasi, and Nadeem sheikh) Moreover, each author declares that this or comparable content has not been submitted to or published in any other publication. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.