

Cancer cell dormancy: An update to 2025
- VNUHCM-US Stem Cell Institute, University of Science, Viet Nam National University Ho Chi Minh City, Viet Nam
- Vietnam National University Ho Chi Minh City, Viet Nam
Abstract
About 70 years ago, scientists first observed groups of cancer cells in a “temporary mitotic arrest,” a dormant state that complicates treatment and increases the risk of recurrence. Recent updates have provided novel insights into the mechanisms driving cancer cell dormancy, especially in relation to how dormant cells evolve and develop resistance to treatments over time. This phenomenon is particularly concerning in breast cancer, where dormant cells can 'wake up' after extended periods, contributing to cancer relapse. Dormancy, akin to hibernation in animals, occurs when cancer cells enter a resting phase (G0/G1 phase) in response to stressors like nutrient deprivation or hypoxia. Key signaling pathways have been identified that regulate the balance between proliferation and dormancy, with some pathways playing critical roles in maintaining dormancy for years. Notably, cancer dormancy has been linked to enhanced stemness and increased resistance to therapies, making drug resistance a significant challenge. Despite promising advancements, existing strategies to target dormant cancer cells have not yet achieved complete eradication, leaving surviving cells that can trigger relapse. A particularly important future direction is the development of combination therapies, which hold potential for preventing recurrence and improving patient outcomes by targeting multiple mechanisms that govern dormancy and reactivation.
Introduction
According to statistics from the Global Cancer Observatory (GLOBOCAN) in 2022, the global incidence of cancer is on an upward trend. Nearly 20 million new cancer cases were reported, and the number of deaths reached approximately 10 million1. Numerous studies have been carried out to identify the underlying causes responsible for the severity and lethality of cancer.
The observation of tumor recurrence in the human body dates back not only to modern times but also to the early centuries A.D. In De medicina, a medical treatise compiled by the ancient Roman encyclopedist Aulus Cornelius Celsus, it was noted that numerous patients experienced the redevelopment of malignant tumors following a period of remission after surgical excision2. In 1934, Rupert Allan Willis first hypothesized that cancer cells stayed silent in tissues where they were localized, as he observed delayed metastases in patients who showed no local recurrence following the removal of their primary tumor3. In 1954, Geoffrey Hadfield proposed that cancer cells, intentionally stalling their activities, entered a phase known as “temporary mitotic arrest”4. Gimbrone et al. (1972) demonstrated that tumor dormancy in vivo can be maintained by preventing neovascularization, highlighting angiogenesis as a critical switch for tumor progression5. This dormant state renders cancer cells difficult to be targeted and eradicated. Recently, using single-cell RNA-sequencing technology, Wang et al. (2022) confirmed that nearly 400 genes exhibit altered expression in dormant cancer cells residing within bone niches6. In addition, a multiomics analysis by Laguillaume et al. (2024) provided insights into the genetic and non-genetic mechanisms governing cancer cell dormancy, highlighting both the complexity and cancer type-specificity of this phenomenon. The identification of both shared and distinct molecular signatures across two models lays a foundation for developing therapeutic strategies aimed at eliminating dormant cells to prevent recurrence and metastasis7. Over time, dormancy allows cancer cells to evolve into populations resistant to current therapies. These findings have established the foundation for oncologists’ understanding of dormant cancer cells as a potential threat to patients when these cells become reactivated (Figure 1).
The concept of cancer dormancy first emerged from German pathologist Rudolf Virchow’s clinical observations in the late nineteenth century, when he noted that some malignant cells survived post-treatment and later gave rise to recurrent tumors with similar histological traits8. In the 1930s and 1950s, early concepts describing non-proliferative or slowly cycling cancer cell populations were proposed3, 4. Gimbrone et al. (1972) and Folkman et al. (1995) summarized that tumor dormancy depends on suppressed angiogenesis, suggesting that reactivation and progression require neovascularization, thereby identifying angiogenesis as a key therapeutic target5, 9. Since the 2000s, in-depth studies have uncovered molecular and cellular mechanisms underlying the dormant state of cancer cells, emphasizing the pivotal roles of microenvironment signals and the immune system, and suggesting potential biomarkers for precision medicine in diagnosis and relapse prevention10, 11, 12, 13.
In this review, we primarily focus on the signaling pathways that lead cancer cells into a dormant state. Also, we discuss how dormant cancer cells are reawakened and examine the similarities between dormant cancer cells and cancer stem cells. Lastly, this review highlights novel strategies targeting dormant cells, which have been explored in in vitro, in vivo, and clinical settings. However, further research is needed to gain a deeper understanding of the mechanisms involved in cancer dormancy, to develop more precise treatment, and to prevent tumor relapse in the future.
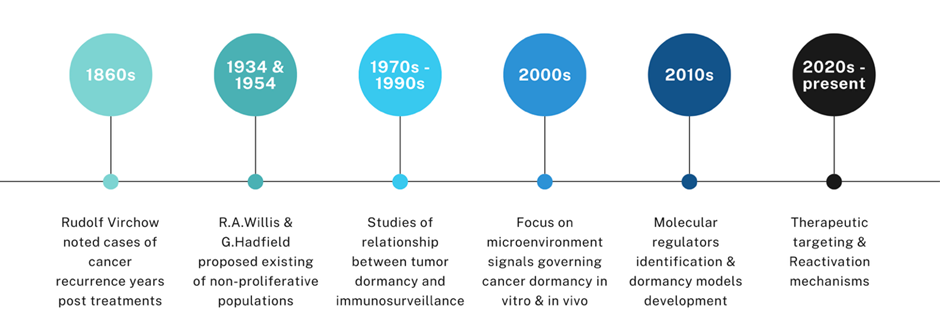
A brief history of studies in cancer dormancy.
Current Understanding of the Mechanisms That Maintain Dormancy in Cancer
After observing dormant populations of cancer cells in breast cancer patients, Retsky et al. (2005) reported that cancer recurrence arises from the reactivation of these dormant cells14. Mechanistically, this unique dormant state mirrors the hibernation mechanism seen in certain animals exposed to harsh conditions in temperate and cold regions15. Cancer cell dormancy can be understood as an intermediary between the G0 and G1 phases of the cell cycle—a “resting” state in which they restrict metabolism and proliferation12, 16. To enter this silent state, stress factors such as oxygen deprivation, limited nutrients, or chemical stress prompt changes in intracellular metabolism, allowing cancer cells to endure adverse conditions without being destroyed12.
Most researchers maintain that activation of the extracellular signal-regulating kinases (ERKs) plays a decisive role in determining whether cells proliferate or enter a dormant state. Additionally, p38, a member of the mitogen-activated protein kinase (MAPK) family, is vital in regulating both normal cell survival and cancer cell behavior17. When excessive cell proliferation is triggered by extracellular ERK signaling, the p38 kinase becomes activated to counterbalance ERK activity, driving the cell into an intermediate state within the G0/G1 phase. The upregulation of ERK and p38 MAPKs is considered the first "golden" signal that influences both cell proliferation and dormancy18. While ERK phosphorylation promotes proliferation, p38 phosphorylation opposes it by inducing cellular dormancy. Therefore, a lower ERK/p38 expression ratio is an indicator of the dormant state in cancer cells. Furthermore, proteins encoded by the F-box and WD repeat domain containing 7 (FBXW7) gene regulate mitotic activity by targeting key proteins such as cyclin E and c-Myc for degradation19, 20, thereby suppressing proliferation and maintaining quiescence21. These findings underscore the importance of cell cycle-regulating signaling pathways in controlling dormancy.
Tumor growth factor beta (TGF-β) stimulates the expression of cell cycle-related and dormancy-related genes in cancer cells such as p15, p21, and p2722. Studies by Bragado et al. (2015) and Sosa et al. (2013) demonstrated that TGF-β2 and all-trans retinoic acid (atRA), derived from the bone microenvironment, can cooperate with intrinsic tumor signals to promote cellular dormancy. These dormant cells are characterized by growth arrest, enhanced survival, and strong expression of pluripotency-associated genes23, 24. Kobayashi et al. (2011) showed that bone morphogenetic protein 7 (BMP-7), secreted by bone stromal cells, induces dormancy in prostate cancer cells via the p38 pathway and upregulation of the metastasis suppressor gene NDRG125. Buijs et al. (2007) highlighted the capacity of BMP-7 to selectively inhibit bone metastasis26. More recently, Nobre et al. (2021) reaffirmed the dormancy-regulating roles of TGF-β and BMP-7 signaling pathways originating from NG2/Nestin mesenchymal stem cells (MSCs)27. Thus, in the bone microenvironment, atRA, TGF-β2, and BMP-7 are supplied in excess, fostering long-term stability and dormancy of cancer cells for years or even decades. Additionally, abscisic acid in bone marrow and serum induces G0 cell cycle arrest in prostate cancer cells via PPARγ signaling28. Prostate cancer cells have also been found to adhere to osteoblasts within the hematopoietic stem cell niche, resulting in TBK1 upregulation, which suppresses mTOR signaling and promotes cellular dormancy and drug resistance29. Furthermore, leukemia inhibitory factor from bone marrow stromal cells inhibits the growth of breast cancer cells and drives dormancy in bone through its ligand and STAT3 signaling30. These findings reveal a complex network of signaling interactions between bone marrow cells and dormant cancer cells.
Uniquely among mechanisms supporting cancer dormancy, breast cancer cells cannibalize MSCs, potentially acquiring a dormant phenotype via TWIST1 and MAPK upregulation31. A hybrid breast cancer cell line, MDA-MSC-hyb5, remained dormant in mouse tissue and, once activated by unknown factors, developed 1.8-fold faster than its parental MDA-MB-231 cells32. Bartosh et al. (2016) hypothesized that key factors of the senescence-associated secretory phenotype become upregulated after MSC cannibalism, inducing a dormant phenotype by spreading growth arrest signals31. Adipose mesenchymal stem cells may promote cancer dormancy and increase chemoresistance via exosomal microRNAs, which are transferred to breast cancer cells in co-culture33. However, interaction with dormant breast cancer cells can trigger tenascin production by adipose-derived stem cells, subsequently heightening cancer invasiveness34. MSCs also contribute to reduced proliferation, enhanced adhesion, and decreased migration of cancer cells via their extracellular vesicles35. These findings clarify the mechanisms that precipitate tumor relapse and metastasis.
PI3K/AKT is a critical biological pathway in cancer cells, given its direct involvement in cell proliferation, survival, and metabolism36. Inhibiting PI3K/AKT is an effective approach to prevent the aggressive growth of various cancer cells37, 38, 39, 40, 41. Endo et al. (2014) observed that colorectal cancer cells entered dormancy when AKT was blocked under hypoxia and limited growth factor availability42. Experiments on different cancer cell types show that impairing the PI3K/AKT signaling pathway upregulates p21 and p2743, 44, 45, 46, two central inhibitors that can modify the cell cycle and halt proliferation.
Epigenetic modifications have recently gained prominence in uncovering the mechanisms of cancer cell dormancy. Several studies highlight their crucial roles across cancer types. For instance, Sun et al. (2022) reported that in head and neck squamous cell carcinoma (HNSCC), TGF-β and p38 signals stimulate macroH2A1 or macroH2A2 overexpression in an autocrine loop, thereby inducing reversible dormancy and preventing metastasis formation47. Likewise, Ferrer-Diaz et al. (2024) suggested that H3K4 methylation is vital for breast cancer stem cell survival, with methyltransferases KMT2B and KMT2D maintaining a dormant and drug-resistant phenotype48. Additionally, ovo-like (OVOL) transcription factors appear to foster dormancy traits in aggressive breast cancer cells, including growth arrest, morphological changes, and reduced migration. OVOL1 and OVOL2 upregulate E-cadherin while downregulating mesenchymal markers—an expression pattern associated with poor prognosis in ERlow breast cancer patients. OVOLs also enhance chromosome 1 open reading frame 116 (C1orf116)—a potential autophagy receptor—to preserve the epithelial phenotype and modulate tumor behavior. The OVOL-C1orf116 axis alters metabolism by lowering glutathione-related metabolites and raising amino acids, indicating a capacity for redox regulation and amino acid recycling during dormancy49. In ER+ mammary carcinoma (ER+ MC) cells, hypomethylation upregulates Trefoil factor 3 (TFF3), sustaining dormancy-related traits and therapy resistance via a BCL2-dependent mechanism50. In parallel, Llinas-Bertran et al. (2024) revealed that extended dormancy in ER+ MC cells induced by tamoxifen or aromatase inhibitors was not linked to genetic mutations but rather to epigenetic alterations, specifically increased heterochromatin marks (H3K9me2, H3K27me3, and H4K20me3)51. Laguillaumie et al. (2024) discovered that dormant melanoma cells keep genetic stability, whereas their leukemia counterparts accumulate mutations during dormancy, implying ongoing genetic evolution. This study also identified distinct CNVs, epigenetic marks, and changes in gene and protein expression—particularly in metabolic and dormancy-associated genes such as Vars2, Eno1, Nudt5, and Capg7. These observations not only pinpoint potential therapeutic targets but also demonstrate clinical relevance through patient-derived data.
Just as some animals hibernate to withstand unfavorable conditions, cancer cells seem to adopt dormancy as a survival strategy. Based on the evidence discussed, it is clear that signaling pathways regulating dormancy in various cancers are highly diverse. When conditions improve or undergo certain changes, these dormant cells reactivate and regain malignancy—they are widely considered a primary cause of cancer recurrence after treatment.
The “awakening” of dormant cancer cells
Several studies have investigated how the reactivation of cancer cells from a dormant state greatly contributes to tumor recurrence, leading to serious complications such as malignant transformation, metastasis, or even death52. Some studies have shown the possibility of carcinogenesis and awakening from dormancy in cancer cells under chronic inflammatory conditions among patients with pre-existing diseases53. Attenuated interferon-gamma (IFNγ) has been shown to facilitate the escape of cancer cells from dormancy and promote relapse54, 55, 56, 57, as IFNγ serves as a key mediator of dormancy induction in cancer sustained by natural killer cells, potentially through the IDO-Kyn-AhR signaling pathway58, 59. Although immune cells, such as CD8+ T cells, can recognize mutant antigens expressed on cancer cells and keep them in a dormant state60, chronic inflammation can reactivate dormant cancer cells via the EMT process and seed them onto new metastatic sites61. Lipopolysaccharide (LPS) maintains inflammation via neutrophil recruitment and causes dormant cancer cells in mice to reenter their cell cycle. In the course of inflammation, NETs are formed and stimulate the resurgence of dormant cancer cells. In vitro, analysis of the mechanism showed that NE and MMP-9, two NET-associated proteases, sequentially cleave laminin. Similar to the former demonstration, this loss of laminin causes dormant cancer cell proliferation via the initiation of integrin α3β1 signaling pathways62.
Multiple signaling pathways and metabolic alterations have been implicated in the reactivation of dormant cancer cells, contributing to tumor recurrence. In HER2-downregulated dormant breast cancer cells, enhanced fatty acid oxidation generates ATP but also leads to reactive oxygen species (ROS) accumulation, subsequently activating NRF2. Stabilized NRF2 restores redox homeostasis and supports nucleotide biosynthesis through thioredoxin reductase and the pentose phosphate pathway, thereby facilitating tumor resurgence63. In mouse models of lung cancer, PMN-MDSCs promote dormancy escape via S100A8/A9 signaling. In vitro, stress hormones such as epinephrine, norepinephrine, cortisol, and serotonin trigger PMN activation under exogenous S100A8/A9 treatment, which in turn initiates myeloperoxidase and several oxidized or hydrolyzed phospholipids. These lipids can upregulate FGFR signaling and drive proliferation of previously dormant cancer cells. Clinically, elevated S100A8/A9 levels in NSCLC patients correlates with an earlier relapse following tumor resection64.
Additional pathways implicated in dormancy escape include NOTCH4-mediated activation of breast cancer stem cells, Hedgehog (Hh)-driven EMT induction, and ATF6-EGF signaling in NSCLC, which promotes angiogenesis and reactivates slow-cycling tumor cells65, 66, 67. Hypoxia-induced LOXL2 expression in dormant MCF-7 cells also facilitates EMT and a phenotypic transition68. Complementing these mechanisms, recent findings demonstrate that exosome-mediated delivery of NR2F1 and its lncRNA regulator NR2F1-AS1 drives the switch from dormancy to proliferation in enzalutamide-resistant prostate cancer. NR2F1-AS1, via interaction with SRSF1, stabilizes NR2F1 expression, leading to hormonal receptor maintenance and sustained proliferative signaling, while HnRNPA2B1 facilitates their packaging into exosomes69. These coordinated metabolic, immunologic, and transcriptional reprogramming events highlight a complex regulatory network underlying cancer dormancy and reactivation.
In general, dormant cancer cells remain quiescent, not growing or expanding. Meanwhile, angiogenesis helps to provide oxygen and nutrients to promote tumor growth. Thus, angiogenesis is considered critical for breaking dormancy in tumors. ARHI is highly expressed on normal ovarian and breast epithelial cells but significantly downregulated or absent in 60-70% of breast and ovarian cancer cases70. ARHI often acts as a tumor suppressor since its re-expression upregulates cell cycle proteins such as p53, p21, and p27, which eventually results in cell cycle arrest in breast cancer cells71. In the highly-expressed-ARHI xenograft model, TIMP3 and CDH1, two anti-angiogenic factors, are upregulated during dormancy since their DNA methylation level decreased. In contrast, increasing methylation leads to the downregulation of these two factors and drives tumor recurrent growth. It is consistent with immunohistochemical staining of PCNA and CD31 results, where they were both reduced in dormant xenografts and escalated in those with progressive growth72. Remodeling of the endosteal niche mediated by osteoclasts can also lead to the reactivation of dormant myeloma cells73, further emphasizing the significance of cell-cell and cell-niche interactions in this process. These insights open new avenues for the development of targeted therapies and the enhancement of treatment efficacy.
Epigenetic regulation plays a critical role in both maintaining cancer cell dormancy and enabling their eventual reactivation. For instance, the histone methyltransferase G9a, which mediates H3K9 methylation, has emerged as a key factor in this process. Under hypoxic conditions, G9a cooperates with hypoxia-inducible factors to repress differentiation genes and activate survival and angiogenic programs, promoting dormancy and tumor growth74, 75. Post treatment, G9a continues to silence pro-inflammatory genes, enabling immune evasion and long-term cellular quiescence76. It also promotes immunosuppression through the Notch pathway and represses adhesion molecules like EpCAM, supporting reactivation and metastasis upon dormancy exit77, 78. Besides, regulators such as EZH1/279, LSD180, HDACs81, and DNMTs82, 83 often interact and form complex networks that control gene expression programs associated with cancer dormancy and reactivation84. Targeting these factors may offer promising therapeutic avenues to prevent cancer relapse in clinical trials.
Awakening cancer cells from dormancy represents a promising area of research for the development of advanced cancer therapies. Based on the above content, we summarized the factors influencing the dormancy and reactivation of cancer cells in Table 1. However, a deeper understanding of this mechanism is essential, and further research is required to translate these findings into clinical practice.
Summary of factors that influence the 'sleep' and 'wakefulness' of dormant cancer cells
|
Factors |
Dormancy maintainance |
Dormancy exit |
|
Signaling |
Upregulation of TGF- 2, BMP-7, p38 MAPK Downregulation of PI3K/AKT |
NOTCH4, ERK, FGFR, integrin |
|
Microenvironment |
Hypoxia, nutrient depletion, bone marrow niches stability |
Chronic inflammation, angiogenesis, uncontrolable microenvironment |
|
Immunity |
IFN |
Downregulated IFN Upregulated NETs Reactivation through MDSCs |
|
Metabolism |
Cell growth arresst Low energy usage |
ROS, NRF2, PPP promote recurrence |
|
Extracellular signals |
Upregulation of TBK, ARHI Downregulation of mTOR |
Upregulation of mTOR or methylation of CDH1/TIMP3 Downregulation of ARHI |
|
Epigenetic regulators |
Histone modifications ( Gene regulation ( |
Enhanced epigenetic-related enzyme activites ( Reprogramming of epigenetic pathways ( |
Dormancy in cancer and cancer stem cells
According to previous studies, cancer cells in a dormant state have been proven to present some properties that resemble cancer stem cells (CSCs), such as resistance to drugs, the ability to evade the immune system, and the capacity to metastasize85, 86. Thus, there may be a complex relationship between cancer stemness and dormancy. Some CSCs remain in a dormant state under nutrient-depleted conditions. BEX2 is significantly expressed in CD274 cholangiocarcinoma cells and maintains these CSCs in dormancy by interacting with the TUFM mitochondrial protein, which eventually leads to mitochondrial dysfunction87. BEX2 is also required for the dormancy of hepatocellular carcinoma88. Colorectal stem-like cancer cells cultured in serum-deprived conditions become dormant because fatty acid oxidation mediates Nanog expression, and Nanog binds to the promoter of p21 and p27 to enhance these cell cycle blockers89. As Nanog plays a vital role in regulating stem cell properties, this relationship between CSC maintenance and dormancy needs to be carefully considered.
Hypoxia plays a pivotal role in both inducing cancer dormancy and maintaining CSCs90, 91, 92, 93, 94, 95, 96. It not only drives cellular quiescence but also reshapes metabolic and signaling pathways critical for tumor progression, immune evasion, and therapeutic resistance97, 98. Chromosome 4 open reading frame 47 (C4orf47), a direct target of HIF-1α, upregulates several cell cycle repressors and downregulates cell cycle promoters to induce G0/G1 arrest and cellular dormancy99. C4orf47 also enhances CD44 expression and sustains CSC-like phenotypes in pancreatic and gallbladder cancers under hypoxia93, 99. Additionally, in salivary adenoid cystic carcinoma, HIF-1α has been recently found to increase DEC2 by lowering miR-922, which possibly influences dormancy via lipid metabolism100. Expanding on these insights, recent studies show that hypoxia-induced metabolic shifts, such as L-2-hydroxyglutarate (L-2HG) accumulation via lactate dehydrogenase activity, preserve stemness and immune evasion in pancreatic cancer101. In triple-negative breast cancer, oxidative ATM activation under hypoxia maintains CSC traits and enhances therapy resistance102. Glioma stem-like cells also upregulate hypoxia-inducible gene 2 (HIG-2) under low oxygen, leading to lipid accumulation and Wnt/β-catenin activation via FZD10, which increases insulin-like growth factor binding protein 2 (IGFBP2) secretion and suppresses radioimmunogenicity103. In renal cell carcinoma, a multi-omics model confirmed that hypoxia modulates stemness and immune infiltration104. Similarly, in head and neck squamous cell carcinoma, hypoxia-driven circulating tumor cells exhibit altruistic behavior and therapy resistance independent of genetic mutations105. Collectively, these findings underscore the multifaceted role of hypoxia in orchestrating dormancy and stemness through metabolic reprogramming, epigenetic regulation, and microenvironmental adaptation, marking it as a central target in overcoming cancer recurrence and resistance. Notably, neoadjuvant bevacizumab improved oxygenation in glioblastoma, reduced CSC characteristics, and enhanced treatment response106.
Autophagy activates when cells are in a state of stress to help cancer stem cells survive under these conditions107. When cancer stem cells enter a quiescent state, they do not proliferate, and autophagy is often highly activated. Dormant MCF-7 breast cancer cells express stem cell markers such as CD44 and ALDH1. MCF-7 breast cancer cells undergo autophagy activation to maintain a dormant status, as autophagy inhibition leads to the reversal of the dormant phenotype108. Alyssa et al. (2019) demonstrated that a high autophagic level reduces Pfkfb3 expression via p62/SQSTM1 and guides breast cancer cells to cellular dormancy109. Because autophagy plays a vital role in controlling the survival of dormant cancer cells, inhibition of autophagic flux results in apoptosis in breast cancer cells, suggesting a therapeutic approach for breast cancer recurrence110. Likewise, autophagy is indispensable for the quiescence of ovarian cancer stem-like cells111. Downregulation of autophagy similarly forces glioblastoma cells to exit dormancy, but this condition also enhances the expression of stemness markers such as Oct-4 and CD133112. The influence of the autophagy process on the expression of stemness markers varies among different cancer types, as autophagy may act as a “double-edged sword” in cancer, with its role in promoting or preventing tumorigenesis still unclear113. However, autophagy strongly affects the promotion of cellular dormancy in general. Harsh conditions within the tumor microenvironment can cause CSCs or dormant cancer cells to accumulate damage, which potentially leads to cell death. Autophagy functions as a protective mechanism that helps manage intracellular stress and stabilize genome integrity by recycling damaged organelles or misfolded proteins114. Autophagy not only keeps CSCs or dormant cancer cells in an inactive state but also enables them to initiate tumor formation when environmental conditions become favorable, allowing them to exit dormancy and re-enter the cell cycle110, 115, 116, 117.
Drug resistance in cancer stem cells and dormant cancer cells
Another aspect that links cancer stem cells with dormant cancer cells is their shared capacity for resistance to drugs and therapies. Drug resistance in cancer stem cells is largely mediated by ATP-binding cassette (ABC) transporter proteins118. These transmembrane transporters are not exclusive to cancer cells, they are also found in various normal cells throughout the body, where they regulate the transport of substances across the cell membrane, contributing to endocrine and exocrine balance. In cancer cells, mutations often occur in the nucleotide-binding domain of ABC transporters, which not only alter drug transport but also impair intracellular energy transfer. As a result, therapeutic agents are unable to exert cytotoxic effects, contributing to drug resistance through ABC transporters119. In addition, metastatic cancer cells infiltrate the lymphatic and vascular systems, disseminating to distant organs before entering a dormant state. During dormancy, they may secrete growth factors and cytokines in preparation for future activation and metastasis formation120. This behavior is considered a key step in the development of metastatic tumors and represents a major cause of cancer-related mortality.
As mentioned above, the ratio between activity of p38 and ERK1/2 is an important indicator in identifying the level of proliferation and dormancy in cancer cells. In particular, p38 acts as a central controller of many pathways related to drug resistance in different types of tumors. SOX9, a downstream signal of p38, has been implicated in drug resistance in breast, esophageal, and cholangiocarcinoma stem cells121, 122, 123. SOX2 and SOX9 are strongly expressed in breast and lung cancer stem cells through DKK1 inhibiting the Wnt signaling pathway, which maintains dormancy in cancer cells124. Although suppression of the Wnt pathway often enhances the therapeutic effect of cancer drugs125, 126, 127, 128, 129, a study in lung cancer cells has also shown that chemotherapy resistance persists even when this pathway is inhibited130. In HEp3 cells, activation of two stress response regulators, protein kinase-like ER kinase (PERK) and BiP, depends on p38 signal. Upregulation of BiP inhibits Bax, an important signal in drug-induced apoptosis, and renders HEp3 cells resistant to several chemotherapeutic drugs131.
In some cases, dormant cancer cells also adjust cellular metabolism to acquire a drug-resistant phenotype. Colorectal dormant cancer cells collaborate with cancer-associated fibroblasts to promote glutamine metabolism and reduce toxicity of 5-fluorouracil (5-FU)132. Glucose starvation could induce massive death of glioblastoma cells under temozolomide (TMZ) or carboplatin treatment, but a small subset of cells escapes and becomes persistent to these chemotherapeutic drugs, entering a quiescent state by enhancing autophagy133. Prostate cancer cells, which are adapted to grow in medium containing the androgen receptor antagonist 2-hydroxy-flutamide, undergo epigenetic modifications. These prostate cancer cells are quiescent and less sensitive to docetaxel, while upregulating pluripotent transcription factors such as Nanog and Oct4. Observations on prostate cancer biopsies confirm that phosphocholine metabolism and methylation are restricted while deacetylation is upregulated. These lead to the reprogramming of prostate cancer cells into cancer stem-like cells134.
These pieces of evidence prove that signaling pathways connecting drug resistance and dormancy in cancer cells are highly complex since dormant cancer cells may use any mechanism to conquer chemotherapeutic agents. However, drug resistance in dormant cancer cells is not always permanent and can be influenced by specific regulatory factors. Kinoshita et al. (2012) demonstrated that reduced expression of nuclear pore protein NUP62 induces cisplatin resistance in dormant ovarian cancer cells, which can be reversed by restoring NUP62 expression135. Ebinger et al. (2016) further demonstrated that dormant leukemia cells cultured ex vivo exhibited reduced drug resistance compared to their previously dormant counterparts in vivo. This finding highlights the potential efficacy of therapeutic strategies that specifically target dormant leukemia cells after their mobilization from the bone marrow niches, which are believed to play a critical role in maintaining cancer cell dormancy136. Therefore, more studies are needed to unveil this association. Dormant cancer cells and cancer stem cells share features like quiescence and drug resistance, yet differ in origin, regulatory pathways, and roles in tumor progression (Table 2).
Comparisons between cancer stem cells and dormant cancer cells
|
Aspects |
Cancer stem cells |
Dormant cancer cells |
|
Proliferation |
Quiescent or slow-cycling, can re-enter cell cycle |
Quiescent (G0/G1 phase), can be reactivated |
|
Drug desistance |
ABC transporters, autophagy, metabolic reprogramming |
p38 signaling, autophagy, niche protection, metabolic shift |
|
Immune evasion |
Low immunogenicity, immune checkpoint regulation |
Secrete factors to evade immunity; reside in protected niches |
|
Hypoxia response |
Maintains stemness; upregulates CD44, Nanog, SOX2, via HIF-1α |
Hypoxia triggers dormancy via HIF-1α, C4orf47, p21/p27 induction |
|
Autophagy role |
Maintains quiescence and stress tolerance |
Maintains dormancy and survival under stress |
|
Angiogenesis |
Produces angiogenic factors to support tumor growth |
Relies on angiogenesis for reactivation and recurrence |
|
Metastasis initiation |
Capable of initiating and maintaining metastases |
Disseminate early and remain dormant before forming metastases |
|
Stemness markers |
CD44, ALDH1, Nanog, SOX2/9, Oct4 |
May express CSC markers during dormancy ( |
|
Metabolic features |
Uses fatty acid oxidation, glucose regulation under stress |
Adapts metabolism for survival ( |
|
Therapeutic implications |
Targeting stemness pathways or ABC transporters |
Targeting dormancy regulators, autophagy, niche exit strategies |
Targeting dormancy in cancer therapies
Currently, treatment options targeting dormancy in cancer focus on three main strategies: (1) maintaining the dormant state of cancer cells, (2) reactivating dormant cells followed by chemotherapy to eliminate them, or (3) completely eradicating these cells while they remain dormant137. Although option (1), which involves peacefully coexisting with dormant cancer cells, carries a high risk due to the potential for uncontrolled reactivation and unforeseen clinical outcomes, options (2) and (3) provide more proactive alternatives. They aim to directly target and eradicate dormant cell populations, thereby significantly minimizing the risk of cancer recurrence in patients.
For type (1), dormant cells must be strictly kept in a “peaceful hibernation” state. Once awakened, they may become more malignant, aggressively increase in population, and metastasize137. Therefore, maintaining dormancy-inducing signals while inhibiting proliferative cues remains a top priority. Tamoxifen, an estrogen receptor antagonist, has been proven to prevent the growth of dormant breast cancer cells and significantly extend patient survival times138. The glutaminase inhibitor CB-839 hinders the reawakening of dormant tumor cells, as recurrent breast cancer cells are sensitive to glutaminase inhibition under the metabolic control of NRF263. In mice, NET-mediated laminin remodeling activates dormant lung cancer cells, but chimeric mouse immunoglobulin G2a antibodies can counteract this process62. Regucalcin sustains dormancy in prostate cancer cells by stimulating p38, downregulating ERK, and reducing angiogenesis through increased miR-23c expression139. Gene therapy strategies targeting uPAR and Src successfully induce a dormancy phenotype in neuroblastoma140 and breast cancer cells141. Targeting the β2-adrenergic receptor in the PMN with ICI-118,551 abrogates norepinephrine-induced S100A8/A9 secretion, preventing dormant tumor cell reactivation64. Palbociclib, a CDK4/6 inhibitor that prevents cells from transitioning from the G0/G1 phase to the S phase, significantly increases survival rates in breast cancer patients. The FDA approved its clinical use for various cancer types in 2016142. Although this strategy is theoretically feasible, it has also been validated in vitro, in vivo, and clinical trials. Recently, certain miRNAs have been identified as key targets for sustaining “peaceful hibernation,” as their inhibition may foster dormancy induction33. Furthermore, epigenetic modification strategies have shown promise. For instance, Clements et al. (2021) demonstrated that histone deacetylase inhibitors reduce proliferation rates in various cancer cell lines, elevate the expression of dormancy-related genes, and delay tumor recurrence143. However, this approach still entails potential risks, as not all cancer cells become truly dormant; some merely exhibit reduced proliferation, which may ultimately trigger tumorigenesis137.
For strategies (2) and (3), several agents are used to disrupt dormancy, making cells more vulnerable to chemotherapy and facilitating their removal. Both in vitro and in vivo data indicate that granulocyte-colony stimulating factor (G-CSF) can reactivate dormant leukemia stem cells, allowing their re-entry into the normal cell cycle. Subsequent therapy with cytarabine or imatinib then effectively eliminates these reactivated cells144. Nimustine and bortezomib have each been shown to decrease dormant breast cancer and myeloma cell populations, respectively, an effect not seen with therapies targeting only proliferating cells73, 145. Combining a Src inhibitor (AZD0530) with a MEK1/2 inhibitor (AZD6244) prevents dormant breast cancer cells from responding to external stimuli, ultimately inducing apoptosis146. Xie et al. (2022) reported that erlotinib and CB-839 effectively kill dormant and drug-resistant colorectal cancer cells132. Moreover, certain genetic modifications in glioblastoma cells reactivate dormant cells and heighten temozolomide-induced apoptosis112. Dwyer et al. (2024) demonstrated that inhibiting autophagy in breast cancer cells, either through pharmacological agents or genetic manipulation, not only postpones tumor recurrence but also diminishes dormant cell survival, as shown in both in vitro and in vivo studies117. Hu et al. (2023) demonstrated that STING pathway activation suppresses the reactivation of dormant metastatic cells in lung adenocarcinoma. Additionally, STING agonist treatment eliminates these cells in a T and NK cell-dependent manner, offering a promising strategy to prevent metastatic relapse147. Growing research is evaluating more compounds for their efficacy in targeting dormant cancer cells, utilizing both in vitro studies and clinical trials (Table 3).
Strategies developed over the past decade (2015-2025) to treat dormant cancer cells
|
Methods or Compounds |
Cancer type |
Outcomes |
Referrence | |
|
Dormancy maintainance |
Indirect or direct eradication of dormant cells | |||
|
Granulocyte-colony stimulating factor (GCS-F) plus cytarabine or imatinib |
Leukemia |
v |
| |
|
Valproic acid |
Breast cancer |
v |
| |
|
Palbociclib |
Various cancer |
v |
| |
|
IFNg plus IDO/AhR inhibitor (Interferon gamma + Indoleamine 2,3-dioxygenase / Aryl hydrocarbon receptor inhibitors) |
Melanoma |
v |
| |
|
Nimustine, bortezomib |
Myeloma |
v |
| |
|
Immunoglobulin G2a antibodies |
Lung cancer |
v |
| |
|
Itraconazole |
Colorectal cancer |
v |
| |
|
Interferon beta (IFN-beta) |
Glioma Breast cancer |
v |
| |
|
FBXW7 ablation |
Breast cancer |
v |
| |
|
ICI-118,551 hydrochloride (ICI-118,551) |
Lung cancer |
v |
| |
|
Exosomal miRNAs |
Various cancer |
v |
| |
|
Glutaminase inhibitor (CB-839) |
Breast cancer |
v |
v |
|
|
Yes-associated protein (YAP) activation |
Lung cancer |
v |
| |
|
Genetics modification |
Glioblastoma |
v |
| |
|
Regucalcin |
Prostate cancer |
v |
| |
|
Targeting enhancer of zeste homolog 1/2 (EZH1/2) |
Leukemia |
v |
| |
|
Histone deacetylase inhibitors (HDAC inhibitors) |
Breast cancer |
v |
| |
|
5-azacytidine plus all-trans retinoic acid (atRA) reprogramming |
HNSCC |
v |
| |
|
uPAR and Scr downregulation via gene therapy |
Neuroblastoma Breast cancer |
v |
| |
|
MacroH2A2 restoration |
HNSCC |
v |
| |
|
Erlotinib |
Breast cancer |
v |
| |
|
Gedatosilib |
Breast cancer |
v |
| |
|
STING activation |
Lung cancer |
v |
| |
|
T-cell immunotherapies |
Breast cancer |
v |
| |
|
Trefoil factor 3 (TFF3) depletion plus CDK4/6 inhibitor |
Breast cancer |
v |
| |
To date, current treatments have not been able to successfully eliminate all dormant cancer cells. Even a minimal population of cells that survive these interventions may later acquire malignant potential, leading to disease relapse and a worsened prognosis for patients. Optimal cancer treatment requires either sustaining dormancy or eliminating these dormant cells, but both strategies remain challenging. Continual therapy to maintain dormancy is often impractical, while reactivating cells to kill them can significantly worsen outcomes if therapies fail157. To address these limitations, a comprehensive understanding of the biological mechanisms governing dormancy and reactivation is essential for developing safer and more effective treatment strategies.
Future directions in cancer dormancy
Understanding and targeting cancer cell dormancy represents a critical frontier in oncology research, particularly in addressing tumor relapse and metastasis that can occur months or even years after initial treatment. Current efforts must focus on elucidating the molecular mechanisms that govern dormancy, including intracellular signaling pathways158, 159, epigenetic regulation160, metabolic reprogramming161, and interactions with the tumor microenvironment159. The development of physiologically relevant in vitro and in vivo models162, 163, along with the identification of robust dormancy-specific biomarkers, is essential for tracking dormant cells and distinguishing them from therapy-induced quiescent populations164. Future directions include developing targeted therapies to either eradicate dormant cells or maintain them in a non-proliferative state indefinitely137, 165, integrating immunological approaches to modulate dormancy surveillance86, and leveraging liquid biopsy and multi-omics technologies for early detection of reactivation events13. In the near future, the establishment of large-scale clinical databases and interdisciplinary collaborations will be pivotal in translating dormancy research into precision medicine strategies166.
Conclusion
The significance of cancer cell dormancy studies has grown markedly because they provide critical insights into many cases of late relapse, metastasis, and therapy resistance. Although dormant cancer cells remain in a non-dividing state, they still possess reproductive capabilities, making them difficult to eliminate with standard therapies, as many are concealed in protective microenvironments167. To build upon new findings, future studies should integrate mechanistic insights (e.g. signaling, epigenetics, metabolic control) alongside advanced techniques such as liquid biopsy and multi-omics to continuously detect and monitor dormancy-related changes in real time. Furthermore, developing reliable dormancy-specific biomarkers and enhanced preclinical models will improve our ability to clearly distinguish dormant cells from therapy-induced quiescent cells. Ultimately, revolutionizing cancer management post-treatment and preventing disease recurrence could be achieved by devising innovative strategies to eradicate or durably contain dormant cells, marking a breakthrough in precision oncology.
Abbreviations
AhR: aryl hydrocarbon receptor, ALDH1: aldehyde dehydrogenase 1, ARHI: aplasia rag homolog member 1, atRA: all-trans retinoic acid, BCL2: B-cell lymphoma 2, BMP-7: bone morphogenetic protein 7, C4orf47: chromosome 4 open reading frame 47, C1orf116: chromosome 1 open reading frame 116, CNV: copy number variation, DNMT: DNA methyltransferase, EMT: epithelial-to-mesenchymal transition, ERK: extracellular signal-regulated kinase, ER+: estrogen receptor positive, EZH1/2: enhancer of zeste homolog 1/2, FBXW7: F-box and WD repeat domain containing 7, FGFR: fibroblast growth factor receptor, HDAC: histone deacetylase, HNSCC: head and neck squamous cell carcinoma, IDO: indolamine 2,3-dioxygenaseIFN-β: interferon beta, Kyn: kynurenine, LOXL2: lysyl oxidase like 2, LPS: lipopolysaccharide, LSD1: lysine-specific demethylase 1, miRNAs: micro ribonucleotide acids, MMP-9: matrix metalloproteinase-9, NETs: neutrophil extracellular traps, NE: neutrophil elastase, NK cell: natural killer cell, NRF2: nuclear factor-like 2, NSCLC: non-small-cell lung cancer, Pfkfb3: 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, PMN: polymorphonuclear neutrophils, PMN-MDSC: polymorphonuclear myeloid-derived suppressor cell, PPARγ: peroxisome proliferator-activated receptor gamma, STAT3: signal transducer and activator of transcription 3, TBK1: TANK binding kinase 1, TFF3: Trefoil factor 3
Acknowledgments
None.
Author’s contributions
Bui Dinh Khan took primary responsibility for the layout and content of this manuscript. Nguyen Thi Yen Nhi, Pham Duy Khuong, and Tran Ngo The Nhan contributed equally to this work. We would also like to thank Phung Van Khai (School of Biomedical Sciences, Faculty of Medicine, Nursing, and Health Sciences, Monash University, Clayton, Victoria, Australia) for providing feedback on the content, correcting spelling and grammatical errors, and improving wording of this manuscript. All figures were illustrated using free tools and templates from Canva. All authors read and approved the final version of the manuscript.
Funding
This paper is funded by University of Science, Vietnam National University Ho Chi Minh City (VNUHCM) under grant number T2024-82.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data. Examples include ChatGPT, NovelAI, Jasper AI, Rytr AI, DALL-E, etc) and AI-assisted technologies in the writing process before submission.
Competing interests
The authors declare that they have no competing interests.