

Peptides from hypothetical proteins of Lactobacillus acidophilus induce IL-4 and IL-10
- Department of Animal Science, University of Ibadan, Nigeria
Abstract
Commensal bacteria used for probiotics usually pose no health risk. However, the functional mechanisms of these probiotics are not fully understood, thereby necessitating new studies such as this. The study aimed to understand the functional mechanisms of microbial probiotics and characterize their uncharacterized hypothetical proteins. In this study, the probiotic Lactobacillus acidophilus genome was explored for antibiotic resistance genes, characterization of hypothetical proteins, and their relationships with cytokine interleukin-4 (IL-4) and cytokine interleukin-10 (IL-10). The genome has an average G+C content of 34.71 and 1,991,579 bp. It has 1,909, 61, and 12 protein-coding sequences, transfer RNA, and ribosomal RNA genes, respectively. Peptides from QHP2 and QHP5 induce IL-4 and IL-10. They are antigenic, nontoxic, and nonallergenic. This study provides insights into better understanding the functional mechanisms of microbial probiotics and lays a solid background for future studies that may focus on sustainable therapeutic feed additives, food supplements, and vaccine development from the hypothetical proteins of Lactobacillus acidophilus.
Introduction
Microbial improvement of dietary supplements has been extensively documented1, 2, 3, and the importance of lactic acid bacteria (LAB) as a source of probiotics in animal and human nutrition cannot be overemphasized4. LAB are characterized mainly by lactic acid production. They are natural microflora in the gastrointestinal tract of animals and humans. These genera include Lactobacillus, Enterococcus, Pediococcus, Tetragenococcus, Vagococcus, Oenococcus, Leuconostoc, Weissella, Carnobacterium, Lactococcus, and Streptococcus5 .
Among all LAB species, the main candidate strain proposed for probiotic potential is the genus Lactobacillus6, 7. Previous findings showed that strains of Lactobacillus (L. ingluviei, L. acidophilus, and L. salivarius) isolated from the gut contents of chickens exhibited strong resistance to acid and bile salt, antibacterial activity, antibiotic tolerance, and high adherence to intestinal epithelial cells. Meanwhile, L. ingluviei and L. salivarius tested in broiler chickens were found to improve gut health by increasing the population of Lactobacillus and Bifidobacterium, with an associated increase in valeric acid and total short-chain fatty acids7. However, further studies to better understand the safety profile and functional mechanisms of probiotics have been strongly recommended4, 7.
The benefits of Lactobacillus probiotic strains in poultry nutrition, including enhanced digestibility, neutralizing enterotoxins, nutrient absorption, immune response, and growth performance, as well as reducing gastrointestinal colonization risks by foodborne pathogens such as Clostridium, Escherichia coli, Salmonella, and Campylobacter, have been documented8, 9, 10. Interestingly, a full understanding of the functional mechanisms, safety profiles, and characteristics of LAB probiotics remains unexplored4, 7. Therefore, understanding the proteins of the probiotic Lactobacillus may help to provide deeper insight into its functional mechanisms and safety profiles. This study was therefore conducted to understand the functional mechanisms of microbial probiotics and characterize their uncharacterized hypothetical proteins vis-à-vis their relationships with cytokine interleukin-4 (IL-4) and and cytokine interleukin-10 (IL-10).
Methods
Nucleotide sequences of the complete reference genome of Lactobacillus acidophilus in the FASTA format were retrieved from the National Center for Biotechnology Information (NCBI) database for further analysis. The accession number of the complete reference genome is CP005926.2. The sequence is publicly available at NCBI. The reference genome was annotated, and proteome comparison analysis was performed to identify similar bacterial genomes in PATRIC. Seven similar bacterial genomes with complete sequencing data were selected from among the several genomes retrieved. The nucleotide sequences of the selected similar genomes were retrieved from PATRIC.
The probiotic potential of the reference and selected similar genomes was determined with iProbiotics, as well as Lactobacillus probiotic prediction and Bifidobacterium, Lactobacillus and other identifier classifications, http://bioinfor.imu.edu.cn/iprobioticsdev/11. Of the specialty genes of the reference genome, three ARG-associated proteins were further explored as well as seven hypothetical proteins. The three-dimensional tertiary structures of the proteins were obtained from AlphaFold 2.0, https://alphafold.ebi.ac.uk/12, while the three-dimensional tertiary structures obtained were validated using the SWISS-MODEL Interactive Workspace (https://swissmodel.expasy.org/assess)13.
The physicochemical properties of the query ARG-associated and hypothetical proteins were determined using ExPASy ProtParam, www.web.expassy.org/protparam14. The subcellular localization of the queried proteins was determined using PSORTb v3.0.2, https://www.psort.org/psortb/15. Protein-protein network interactions of the query ARG-associated and hypothetical proteins were analyzed using STRING16. Immunogenicity, allergenicity and toxicity evaluations of the query proteins were performed using VaxiJen 3.0, https://www.ddg-pharmfac.net/vaxijen3/17, AllercatPro 2.0, allercatpro.bii.a-star.edu.sg18 and ToxDL, http://www.csbio.sjtu.edu.cn/bioinf/ToxDL/19, respectively. Two of the hypothetical proteins (QHP2 and QHP5), which were antigenic, were further investigated. Peptides from these two hypothetical proteins were analyzed for IL-4 and IL-10 prediction using IL-4Pred and IL-10Pred servers20. Thereafter, those found to induce IL-4 and IL-10 were further investigated for immunogenicity, allergenicity and toxicity as previously described.
Results
Characteristics and annotation results of the selected genome
The average G+C content of the genome was 34.71, the total length of the genome was 1,991,579, and there was 1 contig. This reference genome was annotated using the RAST tool kit (RASTtk)21, which assigned it a unique genome identifier of 1579.814. The genome is in the superkingdom Bacteria and is annotated using genetic code 11. Its taxonomy is: cellular organism > Bacteria > Terrabacteria group > Bacillota > Bacilli > Lactobacylates > Lactobacylaceae > Lactobacillus > Lactobacillus acidophilus. The genome has 1,909 protein-coding sequences (CDS), 61 transfer RNA (tRNA) genes, and 12 ribosomal RNA (rRNA) genes, respectively.
The result of the annotation of the reference genome, denoted as 1579cga, revealed that the genome has 412 hypothetical proteins. The database also contained 1,497 proteins that have been assigned functions, including those with Enzyme Commission numbers (481), those with Gene Oncology (400), and those that have been mapped to KEGG pathways (326)22, 23, 24. Two types of protein families are evident: cross-genus protein families and genus-specific families. The genome contains 1,891 proteins that are genus-specific (PLFams), while 1,894 proteins are cross-genus proteins (PGFams), as shown in Supplementary Table 125.

Characteristics and annotation results of
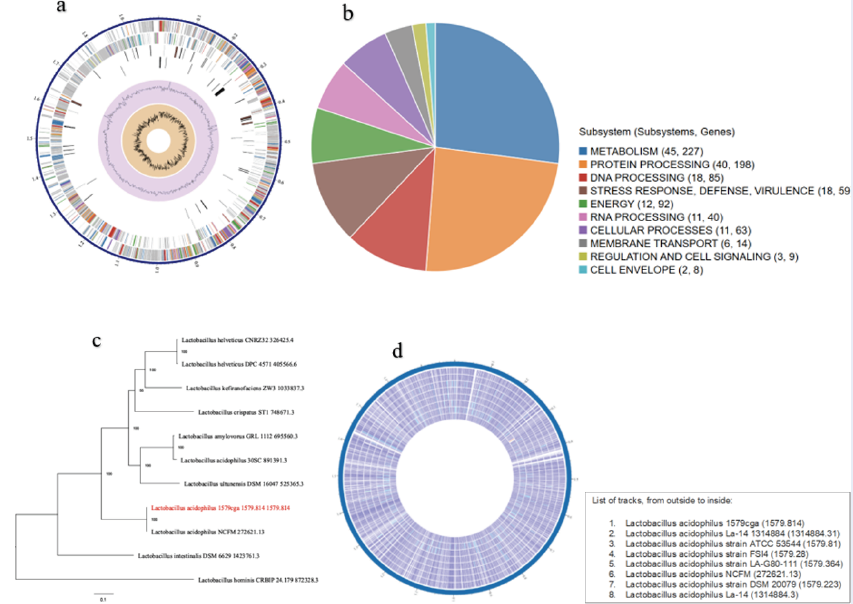
Supplementary Table 2 shows the number of associated genes demonstrating homology to known transporters, virulence factors, drug targets, and antibiotics, as well as the specific source database where homology was found. Figure 1a shows a circular overview of the genome, indicating the contigs, CDSs on the forward strands, CDSs on the reverse strand, RNA genes, CDSs homologous to known antimicrobial resistance genes, CDSs with homology to known virulence factors, GC content, and GC skew. The colors of the CDSs on the forward and reverse strands indicate the subsystem to which these genes belong (Figure 1a).
The unique subsystems associated with this genome are shown in Figure 1b. Ten subsystems were identified as unique to this genome. The associated subsystems include metabolism, protein, DNA processing, stress response, defense and virulence, energy, RNA processing, cellular processes, membrane transport, regulation and cell signaling, and the cell envelope. The subsystems with the greatest number of associated genes were metabolism (45) with 227 genes, followed by protein processing (40) with 189 genes. The least abundant was the cell envelope (2), with 8 associated genes (Figure 1b).
Antimicrobial resistance-associated proteins
Supplementary Table 3 shows an overview of the antimicrobial resistance (AMR) genes annotated in this reference genome as well as the corresponding AMR mechanism. In the PATRIC database, the Genome Annotation Services use a k-mer-based AMR gene detection method, which is based on a curated collection of representative AMR gene sequence variants26, and provides each AMR gene with a functional annotation, broad mechanism of antibiotic resistance, drug class, and, in some cases, a specific antibiotic it confers resistance against. Hence, it should be noted that the presence of AMR-related genes in this genome may not directly imply an antibiotic-resistant phenotype. It may be necessary to consider specific AMR mechanisms and especially the absence or presence of single nucleotide polymorphism mutations that convey resistance.
Representative and reference genomes are manually selected and categorized by the staff of the National Center for Biotechnology Information (NCBI), and such genomes are considered to be of high quality and importance to the research community. On the other hand, PATRIC provides representative and reference genomes that are included in phylogenetic analysis. The closest representative and reference genomes to the genome were identified using Mash/MinHash27 (Figure 1c).
Probiotics prediction scores for reference genome and similar genomes
|
Genome strain |
Probiotic prediction (%) |
|
| ||||
|
|
Probiotics |
Non-probiotics |
Probiotics |
Non-probiotics |
|
|
Other |
|
RF |
99.706 |
0.294 |
3.746 |
96.254 |
99.616 |
0.193 |
0.191 |
|
ATCC |
99.698 |
0.302 |
4.006 |
95.994 |
99.619 |
0.193 |
0188 |
|
DSM 20079 |
99.727 |
0.273 |
3.370 |
96.630 |
99.817 |
0.181 |
0.002 |
|
FS14 |
99.707 |
0.293 |
3.753 |
96.247 |
99.616 |
0.193 |
0.191 |
|
LA1 |
99.692 |
0.308 |
3.739 |
96.261 |
99.618 |
0.194 |
0.188 |
|
La-14 |
99.708 |
0.292 |
3.750 |
96.250 |
99.616 |
0.193 |
0.191 |
|
LA-G80-111 |
99.706 |
0.294 |
3.753 |
96.247 |
99.616 |
0.193 |
0.191 |
|
NCFM |
99.714 |
0.286 |
3.780 |
96.220 |
99.620 |
0.192 |
0.188 |
Evaluation of probiotic potential of selected genomes
The results of the probiotic potential evaluation revealed that all the selected genomes had high probiotic potential, accounting for almost 100% of all the genomes (Table 1). However, the Lactobacillus probiotic prediction scores showed that the percentage of nonprobiotic Lactobacillus was equally high for all the genomes compared with that of the probiotic Lactobacillus. The Lactobacillus, Bifidobacterium, and other classifier scores revealed what is already known, and the genomes were almost totally classified as Lactobacillus (Table 1). A circular view of the proteomic analysis of selected similar genomes of the query reference genome and a circular view of similar genomes whose features are presented in Supplementary Table 4 are shown in Figure 1d and Figure 2.
Circular views of similar genomes of query reference genome for (a)

(A) Three-dimensional tertiary structures of ARG-associated proteins belonging to the reference genome of
Physicochemical properties of query ARG-associated proteins
|
Proteins |
-R |
+R |
#atoms |
Formula |
pI |
#AA |
AI |
MW |
|
RtD1 |
8 |
15 |
3002 |
C991H1531N233O238S9 |
9.61 |
186 |
122.69 |
20806.02 |
|
RtD2 |
17 |
10 |
1573 |
C505H782N132O150S4 |
5.07 |
99 |
86.57 |
11230.80 |
|
RtD3 |
4 |
13 |
1204 |
C396H610N104O92S2 |
10.29 |
70 |
84.86 |
8363.96 |
Physicochemical properties and protein-protein interaction network of ARG-associated proteins
The three ARG-associated proteins are all active, each associated with two roles, namely glycerophosphoryl_diester_phosphodiesterase_(EC_3.1.4.46) and CDP-diacylglycerol--glycerol-3-phosphate_3-phosphatidyltransferase_(EC_2.7.8.5) (Supplementary Table 5). They are all nonantigenic, nontoxic, and nonallergenic. RtD1 (fig|1579.814.peg.642) and RtD2 (fig|1579.814.peg.483) are cytoplasmic, while RtD3 (fig|1579.814.peg.484) is found in the cytoplasmic membrane. The three-dimensional tertiary structures and Ramachandran plots of ARG-associated proteins belonging to the reference genome of Lactobacillus acidophilus are presented in Figure 3.
The total number of negatively charged residues (Asp + Glu) of the proteins ranged from 4 (RtD3) to 17 (RtD2), while the total number of positively charged residues (Arg + Lys) ranged from 10 (RtD2) to 15 (RtD1). The molecular weight was highest for RtD1 (20806.02), while the lowest value was obtained for RtD3 (8363.96). The theoretical pI ranged from 5.07 (RtD2) to 10.29 (RtD3). The aliphatic index ranged from 84.86 (RtD3) to 122.69 (RtD1) (Table 2). The extinction coefficient was very high for all the proteins except for RtD1. RtD1 had a negative grand average of hydropathicity (GRAVY) score, while the rest had positive GRAVY values. The instability index scores were less than 40 for all the query proteins (Table 3).
Physicochemical properties of query ARG-associated proteins (cont’d)
|
Proteins |
GRAVY |
II |
EC |
EC** |
Mammalian reticulocytes, |
Yeast, |
|
|---|---|---|---|---|---|---|---|
|
RtD1 |
0.873 |
17.91 |
40575+ |
40450 |
30 hours |
>20 hours |
>10 hours |
|
RtD2 |
-0.272 |
31.06 |
4470* |
30 hours |
>20 hours |
>10 hours | |
|
RtD3 |
-0.246 |
20.85 |
9970 |
30 hours |
>20 hours |
>10 hours |
The functional partners associated with RtD1 are LBA0663 (0.948), cdsA (0.944), acmA (0.829), fabG (0.821), LBA0660 (0.787), LBA0661 (0.787), LBA0659 (0.732), LBA0657 (0.728), recA (0.678), and ymdA (0.643). The functional partners for RtD2, with their corresponding confidence scores, included LBA0497 (0.771), LBA0496 (0.584), LBA0495 (0.584), LBA0494 (0.572), glpF (0.540), glpF-2 (0.540), glpK (0.519), and glpK-2 (0.519). No change was detected for RtD3 (Figure 3c).
Amino acid composition of the query hypothetical proteins
The hypothetical query proteins are composed of proteins with different amino acid lengths and compositions. Supplementary Figure 1a–g shows the amino acid composition of each of the query proteins. Lys and Leu are the most prominent amino acids of QHP1, while Met, Val, and Phe are the least prominent amino acids in QHP1 (Supplementary Figure 1a). The most prominent amino acids in QHP2 are Leu, Asp, and Ile, whereas the least prominent amino acids in QHP2 are Trp and Cys (Supplementary Figure 1b). Ile, Pro, and Leu are the most abundant amino acids of QHP3, and His, Met, Thr, and Trp are the least abundant amino acids of QHP3 (Supplementary Figure 1c). Gln and Leu are the prominent amino acids, and Arg, Gly, and Phe are the least abundant amino acids in QHP4 (Supplementary Figure 1d). The most prominent amino acids of QHP5 are Lys and Asp, while Cys and Tyr are the least prominent amino acids in QHP5 (Supplementary Figure 1e). Ser and Thr are the most prominent amino acids of QHP6, while Trp and Met are the least prominent amino acids of QHP6 (Supplementary Figure 1f). Lys and Glu are the most prominent amino acids of QHP7, while Cys and Met are the least prominent amino acids of QHP7 (Supplementary Figure 1g).
Subcellular localization scores and prediction for query hypothetical proteins
|
|
Localization scores |
| |||
|
Proteins |
Cytoplasmic |
Cytoplasmic membrane |
Cell wall |
Extracellular |
Prediction |
|
QHP1 |
7.50 |
1.15 |
0.62 |
0.73 |
Cytoplasmic |
|
QHP2 |
7.50 |
1.15 |
0.62 |
0.73 |
Cytoplasmic |
|
QHP3 |
2.50 |
2.50 |
2.50 |
2.50 |
Unknown |
|
QHP4 |
2.50 |
2.50 |
2.50 |
2.50 |
Unknown |
|
QHP5 |
0.00 |
3.33 |
3.33 |
3.33 |
Unknown |
|
QHP6 |
0.32 |
9.55 |
0.12 |
0.01 |
Cytoplasmic membrane |
|
QHP7 |
2.50 |
2.50 |
2.50 |
2.50 |
Unknown |
Subcellular localization and secondary properties of the query hypothetical proteins
The subcellular localization scores of the hypothetical query proteins are presented in Table 4. QHP1 and QHP2 are found in the cytoplasm; QHP3, QHP4, QHP5 and QHP7 have unknown locations; and QHP6 is found in the cytoplasmic membrane. Supplementary Table 7 presents the secondary structure characteristics of the query hypothetical proteins, and Supplementary Figure 2a-g shows a pictorial representation of the helices, sheets, turns and coils of the query hypothetical proteins. The alpha helices (%) ranged between 33.21 (QHP6) and 91.38 (QHP4), the extended strands ranged between 2.53 (QHP1) and 26.64 (QHP6), the beta turns ranged between 1.72 (QHP4) and 8.53 (QHP2), and random coils ranged between 6.90 (QHP4) and 46.04 (QHP7) (Supplementary Table 7).

(A) The three-dimensional structures of the query hypothetical proteins of QHP1 (a), QHP2 (b), QHP3 (c), QHP4 (d), QHP5 (e), QHP6 (f) and QHP7 (g); (B) Ramachandran plots obtained for the three-dimensional structural validation of query hypothetical proteins of QHP1 (a), QHP2 (b), QHP3 (c), QHP4 (d), QHP5 (e), QHP6 (f) and QHP7 (g).
The three-dimensional structures and Ramachandran plots of the query hypothetical proteins
The three-dimensional tertiary structures of the query hypothetical proteins were obtained using Alphafold and viewed using Jmol. The quality of the tertiary structures obtained was further evaluated using PDB files of the retrieved structures in the SWISS-MODEL Interactive Workspace, which yielded Ramachandran plots and other statistics. The three-dimensional tertiary structures, Ramachandran plots and statistics are presented in Figure 4A-B and Supplementary Table 8, respectively. The Ramachandran plots validated the structures obtained as the majority of the residues fell in the (B, A, L) areas (Figure 4B). The favoured Ramachandran areas (%) are 98.70 (QHP1), 98.05 (QHP2), 97.93 (QHP3), 100.00 (QHP4), 95.41 (QHP5), 94.49 (QHP6) and 81.82 (QHP7). The outliers (%) are 0.00 (QHP1‑QHP4), 0.92 (QHP5), 0.37 (QHP6) and 5.45 (QHP7). Only QHP6 had a transmembrane segment. The QMEAN score ranged between ‑4.33 (QHP7) and 1.90 (QHP4), the C_b interaction energy ranged between ‑0.82 (QHP3) and 3.96 (QHP4), all atom pairwise energy values ranged between ‑0.10 (QHP3) and 4.80 (QHP4), the solvent energy values ranged between ‑0.64 (QHP7) and 3.24 (QHP4), and the torsion angle energy ranged between ‑4.30 (QHP7) and 1.22 (QHP4) (Supplementary Table 8).
The immunogenicity, allergenicity and toxicity results of the query hypothetical proteins
The results of immunogenicity, allergenicity and toxicity revealed that nearly all the proteins are safe for consumption and may not have any serious effects on human or animal health. QHP2 and QHP5 were antigenic, and the remaining queried proteins are nonantigenic. All the hypothetical query proteins were nonallergenic (Supplementary Table 9).
Physicochemical properties of query hypothetical proteins
|
Proteins |
-R |
+R |
# atoms |
Atomic composition |
pI |
#AA |
AI |
MW |
|
QHP1 |
12 |
13 |
1302 |
C:403, H:656, N:112, O:130, S:1 |
7.91 |
79 |
87.72 |
9182.37 |
|
QHP2 |
35 |
28 |
4082 |
C:1285, H:2045, N:347, O:396, S:9 |
5.31 |
258 |
94.07 |
28980.00 |
|
QHP3 |
20 |
24 |
3201 |
C:1041, H:1590, N:278, O:287, S:5 |
9.17 |
195 |
82.05 |
22752.07 |
|
QHP4 |
21 |
12 |
1870 |
C:580, H:933, N:153, O:201, S:3 |
4.36 |
116 |
87.50 |
13361.87 |
|
QHP5 |
20 |
15 |
1732 |
C:545, H:852, N:154, O:177, S:4 |
5.46 |
111 |
64.23 |
12521.93 |
|
QHP6 |
29 |
37 |
4422 |
C:1398, H:2220, N:368, O:430, S:6 |
9.32 |
274 |
84.67 |
31255.57 |
|
QHP7 |
28 |
47 |
3421 |
C:1072, H:1713, N:323, O:311, S:2 |
9.82 |
202 |
65.10 |
24166.49 |
Physicochemical properties of the query hypothetical proteins
Table 5 shows the physicochemical properties of the selected hypothetical proteins of the reference genome. The total number of negatively charged residues ranged from 12 to 35, while the total number of positively charged residues ranged from 12 to 47. The total number of atoms ranged between 1302 and 4422. The pI ranged between 4.36 and 9.82, the molecular weight ranged between 9182.37 and 31255.57, and the aliphatic index ranged from 64.23 to 94.07. The instability indices of QHP1, QHP2 and QHP5 were less than 40; hence, they are stable in nature. QHP3, QHP4, QHP6 and QHP7 are unstable proteins. The estimated half-life was the same for all the tested proteins. The atomic formulas for the selected hypothetical proteins were C403H656N112O130S, C1285H2045N347O396S9, C1041H1590N278O287S5, C580H933N153O201S3, C545H852N154O177S4, C1398H2220N368O430S6 and C1072H1713N323O311S2 for QHP1, QHP2, QHP3, QHP4, QHP5, QHP6 and QHP7, respectively.
The presence of Cys, Trp, and Tyr residues is indicated by a high extinction coefficient of QHP2 (22015) but remained unchanged for QHP5 (2980) and QHP7 (16390). The aliphatic indices ranged between 65.10 (QHP7) and 94.07 (QHP2), indicating thermal stability over a wide temperature range. The GRAVY indices were low, ranging between ‑1.299 (QHP7) and ‑0.307 (QHP2). A low GRAVY value is an indication that a protein is hydrophilic (Table 5 ).

Protein-protein interactions of the query hypothetical proteins as predicted by STRING for QHP1 (a), QHP2 (b), QHP3 (c), QHP4 (d), QHP5 (e), QHP6 (f) and QHP7 (g)
Protein–protein interaction network of the query hypothetical proteins
The functional partners for QHP1 are LBA0378, recM, LBA0383, LBA0384, rsml and LBA0386. Both LBA0378 and recM had confidence values of 0.786. The functional partners of QHP2 included LBA0435, LBA0437, LBA0434, LBA0433, pbpX, mprF, pbpX‑2, LBA0432, nagB and LBA0351. LBA0435, LBA0437, LBA0434 and LBA0433 had confidence scores of 0.951, 0.926, 0.895 and 0.854, respectively. QHP3 exhibited functional interactions with LBA0544, LBA0542, LBA0543, cadA, LBA0388, gpmA, LBA0341 and LBA0546, with confidence scores of 0.608 for LBA0544, 0.602 for LBA0542 and 0.602 for LBA0543. The functional partners with corresponding scores for QHP4 were LBA1589 (0.886), LBA1590 (0.881), LBA1591 (0.879), pbpF (0.863), LBA1278 (0.754), LBA0428 (0.744), LBA1594 (0.686), LBA0420 (0.640), ezrA (0.602) and LBA0740 (0.591). The functional partners of QHP5 included LBA1586, trmB, LBA1584, LBA1585, ribT and prtM, and their respective scores were 0.780, 0.556, 0.556, 0.556, 0.537 and 0.522. yycH (0.985), LBA0082 (0.932), LBA0079 (0.908), htrA (0.831), LBA0078 (0.776), LBA1584 (0.640), LBA0740 (0.635), LBA1823 (0.629), ribT (0.627) and LBA0824 (0.619) were the functional partners of QHP6. There were no functional partners found for QHP7 based on the specified conditions, except at lower stringency for which the highest scores were less than 0.4 (Figure 5).

Active sites of the query hypothetical proteins, QHP1 (a), QHP2 (b), QHP3 (c), QHP4 (d), QHP5 (e), QHP6 (f) and QHP7 (g), as predicted by PSORT, showing the active sites (red spot) and amino acid residues in the active sites (shaded portions)
Active site analysis result of the query hypothetical proteins
The results of the active site analysis of the selected hypothetical proteins are presented in Figure 6a-g: QHP1 (a), QHP2 (b), QHP3 (c), QHP4 (d), QHP5 (e), QHP6 (f) and QHP7 (g). The six amino acid residues (Ile, Gln, Lys, Tyr, Ala and Ile) in the active site of QHP1 (shaded) are shown in Figure 6a. The active site of QHP2 contains 36 amino acid residues (Asp, Leu, Asp, Gly, Tyr, Arg, Gly, Lys, Asn, Thr, Asn, Asn, Thr, Arg, Ile, Lys, Met, Asn, Ala, Asp, Met, Asn, Leu, Pro, Ala, Glu, Leu, Asn, Lys, Asp, Asn, Asp, Thr, Asp, Gly and Ile) (shaded in Figure 6b). There were 24 amino acid residues (Ile, Gly, Gln, Ala, Pro, Gly, Ile, Ala, Lys, Lys, Phe, Trp, Asp, Asp, Ser, Ile, Pro, Pro, Gly, Asp, Leu, His, Ser and Arg) in the active site of QHP3, which are shaded (Figure 6c).
QHP4 has five amino acid residues (Ile, Ser, Ala, Leu and Tyr) in its active site (shaded, Figure 6d). There were 35 amino acid residues (Leu, Val, Phe, Ala, Ala, Phe, Val, Ser, Cys, Leu, Tyr, Ile, Lys, Glu, Gly, Arg, Phe, Ala, Leu, Ala, Ala, Ser, Leu, Ile, Met, Phe, Tyr, Ser, His, Ile, Ile, Val, Met, Gln and Ser) in the active site of QHP5 (shaded, Figure 6e). Six amino acid residues (Tyr, Val, Arg, Val, Arg and Ile) were detected in the active site of QHP6 (shaded inFigure 6f). QHP7 contains 90 amino acid residues (Lys, Phe, Glu, Lys, Glu, Gln, Pro, Leu, Arg, His, Thr, Lys, Lys, Asp, Tyr, Leu, Gln, Tyr, Ser, Ser, Glu, His, Leu, Arg, Leu, Lys, Gly, Phe, Asp, Ala, Ala, Arg, Thr, Ser, Ile, Asp, Lys, Lys, Ala, Asp, Tyr, Glu, Tyr, Gln, His, Gln, Gln, Gln, Lys, Ile, Arg, His, His, Arg, Gln, Asp, Asn, Asn, Pro, Phe, Lys, Lys, Arg, Arg, Val, His, Gln, Thr, Phe, Lys, Lys, Lys, Gln, Ser, Ile, Ala, Val, Lys, Lys, Val, Gly, Ser, Phe, Ile, Ile, Lys, Lys, Lys and Arg) in its active site (shaded) (Figure 6g).
Two groups of peptides were derived from each of the two hypothetical proteins found to be antigenic. Peptides from both proteins induced IL‑4 and IL‑10. However, only peptides that induced IL‑4 and IL‑10 and were found to be immunogenic were reported in this study (Supplementary Table 10). QHP2 had more peptides which passed the three tests, while QHP5a had two peptides that scaled through but none were found to pass the three tests in QHP5b. QHP2a and QHP5a were derived from the start of the proteins, while QHP2b and QHP5b were derived from the end parts of the proteins. The derived peptides and their mutants were investigated, and the results are presented in Supplementary Table 10.
Discussion
The importance of studies such as this cannot be overemphasized. The increased risk of antibiotic resistance genes (ARGs) to human health has been attributed to the use of antibiotics28, 29, resulting in an increase in probiotic use, but little is known about their functional mechanisms, necessitating studies like this.
In the present study, only RtD1 had a positive GRAVY value, and the other two proteins had negative GRAVY scores. Proteins with positive GRAVY values are hydrophobic, while those with negative GRAVY scores are hydrophilic14. The instability index scores for the three query proteins were lower than 40; hence, they are all stable in nature. The aliphatic index values are generally high for all the query proteins, indicating that the query proteins are stable over a wide range of temperatures14. RtD2 obtained the lowest extinction coefficient score, which is due to the protein’s lack of any Trp residues.
LBA0663 is a putative transcriptional regulator; cdsA is a phosphatidase cytidyltransferase; COG0575 is a CDP-diglyceride synthetase; acmA is an N-acetylmuramidase; COG1705 is a muramidase (flagellum-specific); fabG is a 3-oxoacyl-(acyl-carrier protein) reductase, while LBA0660 is a putative protease; and COG0612 is a predicted Zn-dependent peptidase. LBA0497 is a hypothetical protein; LBA0495 is a putative phosphoglycerate mutase; LBA0496 is a putative phosphoglycerate mutase, belonging to the phosphoglycerate mutase family; while LBA0494 is a putative surface exclusion protein.
LBA0378, recM, LBA0383, LBA0384, rsml, and LBA0386 are the functional partners of QHP1. LBA0378 is a hypothetical protein that may bind to DNA and alter its conformation. It may be involved in the regulation of gene expression. recM is a recombinational DNA repair protein that may play a role in DNA repair. LBA0435 and LBA0433 are hypothetical proteins; LBA0437 is a COG4478 predicted membrane protein; and LBA0434 is a putative UDP-sugar hydrolase of the 5′-nucleotidase family. LBA0543 is a hypothetical protein; LBA0555 is a transcriptional regulator; and LBA0542 is a putative heavy-metal-transporting ATPase. LBA1589 is a CMP-binding factor. LBA1590 and LBA1278 are hypothetical proteins; LBA1591 is a phosphoesterase and a COG0420 DNA repair exonuclease, while pbpF is a penicillin-binding protein.
LBA1586 is a histidine triad HIT family protein. yycH and LBA0082 are hypothetical proteins. LBA0079 is a putative histidine kinase; htrA is a putative heat shock–related serine protease; and LBA0078 is a VicR (COG0745), a response regulator conserved with a CheY-like receiver domain and a winged-helix DNA-binding domain. Many proteins perform their functions independently. However, several of these proteins also interact with other proteins for proper biological activity. Hence, characterizing protein-protein interactions is key to understanding protein functions30.
The market for probiotics, which are primarily used as dietary supplements, cosmetics, and medicines, is increasing annually. It has been reported that this market increased from USD 66.9 billion in 2022 to USD 73.14 billion in 2023, with a compound annual growth rate (CAGR) of 9.3% and a forecast growth of 3.75% on average until 202631. Considering this enormous market value, which in part has been attributed to increasing personal efforts toward individual health, there is a need for concerted efforts to ensure its safety via a holistic and multidimensional approach. Interestingly, most commercial probiotics are from the genus Lactobacillus, which has been reclassified into approximately 25 genera32, and bifidobacteria.
Probiotics are live microorganisms that confer health benefits to the host when consumed or applied in an adequate amount, although this definition has been amended to include “a defined content, an appropriate number of viable cells at the end of the product’s shelf life and adequate evidence of health benefits, as well as being safe for their intended use”5. Therefore, for probiotics to be safe for intended use, they should contain viable microorganisms within their shelf life, and their strains must be adequately and clearly characterized33. They must also be free from contamination34. To ensure the safety of dietary products, they must be free from antibiotic resistance genes35, although studies linking ARGs with probiotic strains are rare36, 37, which demands that recommended probiotics for animal feeding, food supplementation, cosmetic, or therapeutic purposes be free from antibiotic resistance. In 2019, approximately 1.27 million human deaths were attributed to antibiotic-resistant bacteria38, which implies that antimicrobial resistance is a severe global health problem39, 40. It has been reported that ARGs and their hosts are serious global threats, and are estimated to be responsible for approximately 700,000 annual human deaths globally, which may reach 10 million by 205041. Awareness of food animal safety, free from chemical and antibiotic contamination, is increasing, necessitating relentless efforts to provide safe and sustainable alternatives42, 43.
The present study reported that the Lactobacillus acidophilus reference genome is associated with antibiotic resistance genes (psgA and GdpD-family proteins), which are predicted to be resistant to daptomycin. Interestingly, it has been suggested that ARGs from consumed probiotics can be transferred to human gut bacteria31, and the same may be true for animals. As expected, the transfer of ARGs to other bacteria may lead to a decrease in the effectiveness of antibiotic treatment37. Growing evidence of gene exchange between pathogenic strains and beneficial commensal bacteria in the intestinal tract can help beneficial bacteria become reservoirs of antimicrobial resistance38. However, being an in silico analysis, we strongly recommend that further studies adopting an experimental approach be conducted to ascertain this claim.
Three mechanisms through which the horizontal transfer of resistance genes can occur have been identified: transformation, transduction, and conjugation. In transformation, foreign genetic material is obtained from the extracellular environment44, while in the transduction mechanism, parts of the bacterial DNA are incorporated into the bacteriophage during replication, which subsequently infects another bacterial cell, thereby causing the transfer of genetic material45. During the process of conjugation, cell-to-cell contact induces DNA transfer44.
Daptomycin is a relatively new antibiotic used to treat infections caused by Gram-positive bacteria and as a replacement for antibiotics to which bacteria have developed resistance. It kills microorganisms by rapid membrane depolarization, disruption of DNA, and loss of membrane potential, as well as by inhibiting RNA and protein synthesis46. Findings from a relatively recent study showed that daptomycin resistance was low in staphylococci, and the authors recommended that the antibiotic can still be used for the treatment of staphylococcal infections worldwide47. However, resistance to this antibiotic may be concerning, as research findings are increasing. Hence, we recommend further studies to investigate this assertion.
In this study, peptides derived from QHP2 and QHP5 induced IL-4 and IL-10, which has two benefits: providing insights into the functional mechanisms through which microbial probiotics enhance the host’s immune response (i.e., probiotics improve host immunity by inducing IL-4 and IL-10) and indicating that the derived antigenic, nonallergic, and nontoxic peptides that induce IL-4 and IL-10 may be used as potential candidates for vaccine development, therapeutic feed additives, and food supplements. Peptides that induce anti-inflammatory cytokines such as IL-4 and IL-10 are considered anti-inflammatory20.
Cytokines are known as a broad group of glycoproteins or soluble proteins with low molecular weight (ranging from 6 to 70 kDa), produced transiently in response to various biological stimuli. They can be produced by nearly every cell type and affect virtually all main cellular processes. They play crucial roles in orchestrating cell-to-cell communication and biological functions48. They bind to specific transmembrane and membrane-anchored receptors in target cells and activate downstream intracellular signaling cascades that lead to gene expression modulation49, 50. They play key roles in the regulation of many physiological processes (stem cell differentiation, apoptosis, cytoskeletal organization, embryonic development, cell proliferation, activation, migration, wound healing, and survival)50, 51. They regulate innate and adaptive immunity, coordinating humoral, cytotoxic, and cellular immune responses, mediating communication between immune and nonimmune cells, controlling immune cell trafficking and tissue organization, affecting the microenvironment, and regulating inflammation48, 52.
The roles of IL-4 and IL-10 in the immune system cannot be overemphasized. They play crucial roles in biological processes. For instance, IL-4 has been reported to play a significant role in allergic responses. It controls immunological responses, increases IgE production, is involved in antibody isotype switching, and can block negative effects of Th1. It has strong anti-apoptotic properties, having a great impact on a range of target cells53. IL-4 has anti-cancer and protective effects in neurologic disorders54, 55. It can improve memory, reduce inflammation related to psoriasis in humans, ameliorate arthritis56, and help control immunological responses57.
A relationship exists between IL-4 production and several pathogen responses58. The cellular immune response to disease is mediated by the processing and presentation of antigens on cells by the major histocompatibility complex (MHC). Before extrinsic antigens are presented and processed by MHC class II, they are first treated in the lysosome. The production and secretion of a cytokine pattern therefore occur when the peptides carried by MHC class II engage and bind with CD4 T cells57. The cytokines released (Th1, Th2, Th17, or iTregs) influence the differentiation of T-helper cells into several T-cell populations59. IL-4 has a tendency to stimulate CD4 T cells to promote Th2 cell differentiation during T-cell activation. Antigen-presenting cells proliferate and differentiate via this cytokine57, 58, 59, 60.
IL-4 is important for mediating allergic inflammation, having the ability to counteract pro-inflammatory immune responses triggered by Th1. It can restrict the production of pro-inflammatory cytokines and may successfully increase cytolytic T-cell activity in vitro and encourage T-cell proliferation, aiding CD8 cell growth. It plays crucial roles in many immune cells and controls macrophage phenotypes, which in turn mediate and influence tissue healing and homeostasis61.
IL-10 is an endogenous “danger signal” released in response to the peak of circulating pro-inflammatory cytokines, aiming to protect the organism from harm caused by a hyperinflammatory state62, 63. Carlini et al.64 noted that IL-10 mediates innate and adaptive immunity, having a multifaceted nature in stimulating or inhibiting important immune pathways. As an immune modulator, it can decrease detrimental inflammation, inhibit cancer progression, and curb disease conditions.
Conclusions
This study provides insights into a better understanding of the functional mechanisms of microbial probiotics, suggesting that microbial probiotics enhance the host’s immune response by inducing IL-4 and IL-10. The characterization of uncharacterized hypothetical proteins of the L. acidophilus genome revealed their physicochemical properties, subcellular locations, structures, and interactions with other proteins, and this study lays a solid background for future research that may focus on vaccine development from these hypothetical proteins of L. acidophilus, as well as on therapeutic studies of microbial probiotics in general. Being an in silico analysis, the authors strongly recommend further studies adopting experimental analyses involving animal trials—such as stimulating macrophage/T cell lines with recombinant QHP2/5 and checking IL-4 and IL-10 mRNA expression—as these may be necessary.
Abbreviations
AMR – antimicrobial resistance; ARG – antibiotic resistance genes; bp – base pairs; CAGR – compound annual growth rate; CDS – coding sequences; G+C – guanine + cytosine content; GRAVY – grand average of hydropathicity; IL-4 – interleukin-4; IL-10 – interleukin-10; IL-4Pred – name of IL-4 prediction server; IL-10Pred – name of IL-10 prediction server; KEGG – mentioned as “KEGG pathways”; L. (e.g., L. acidophilus) – short for Lactobacillus; LAB – lactic acid bacteria; MHC – major histocompatibility complex; NCBI – National Center for Biotechnology Information; PATRIC – name of the database; QHP1…QHP7 – labels for “query hypothetical proteins”; RASTtk – RAST tool kit; rRNA – ribosomal RNA; tRNA – transfer RNA.
Acknowledgments
Olawumi Adejumo, Mrs. Evelyn and Mr. Sunday Ehimare are appreciated for their support during data curation.
Author’s contributions
IOA and OAA reviewed and edited the manuscript, IOA designed the study, performed data analysis and wrote the first draft. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
The datasets used in this manuscript are included and where not, adequate links are provided.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.